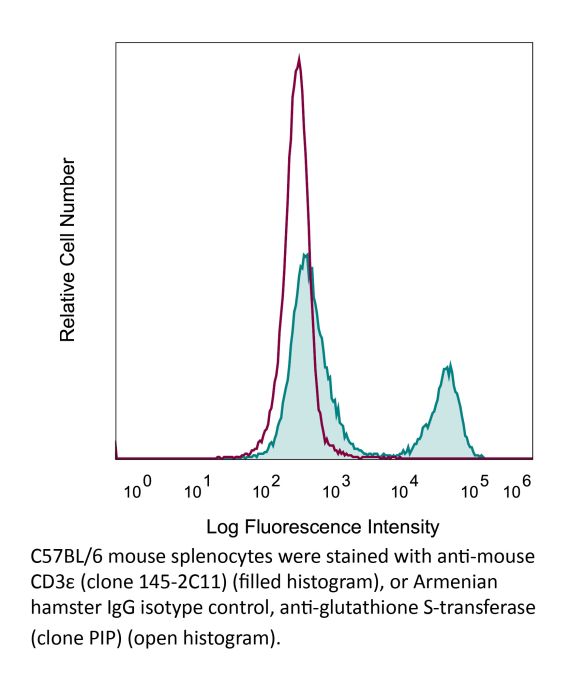
InVivoMAb anti-mouse CD3ε
Product Description
Specifications
| Isotype | Armenian Hamster IgG1 |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse BM10-37 cytotoxic T cells |
| Reported Applications |
in vivo T cell depletion in vitro T cell stimulation/activation Immunofluorescence Flow cytometry Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein A |
| RRID | AB_1107634 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Tang, W., et al (2014). "The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells" Immunity 41(4): 555-566.
PubMed
Bcl-3 is an atypical member of the IkappaB family that modulates transcription in the nucleus via association with p50 (NF-kappaB1) or p52 (NF-kappaB2) homodimers. Despite evidence attesting to the overall physiologic importance of Bcl-3, little is known about its cell-specific functions or mechanisms. Here we demonstrate a T-cell-intrinsic function of Bcl-3 in autoimmunity. Bcl-3-deficient T cells failed to induce disease in T cell transfer-induced colitis and experimental autoimmune encephalomyelitis. The protection against disease correlated with a decrease in Th1 cells that produced the cytokines IFN-gamma and GM-CSF and an increase in Th17 cells. Although differentiation into Th1 cells was not impaired in the absence of Bcl-3, differentiated Th1 cells converted to less-pathogenic Th17-like cells, in part via mechanisms involving expression of the RORgammat transcription factor. Thus, Bcl-3 constrained Th1 cell plasticity and promoted pathogenicity by blocking conversion to Th17-like cells, revealing a unique type of regulation that shapes adaptive immunity.
-
Tang, W., et al (2014). "The oncoprotein and transcriptional regulator Bcl-3 governs plasticity and pathogenicity of autoimmune T cells" Immunity 41(4): 555-566.
PubMed
Bcl-3 is an atypical member of the IkappaB family that modulates transcription in the nucleus via association with p50 (NF-kappaB1) or p52 (NF-kappaB2) homodimers. Despite evidence attesting to the overall physiologic importance of Bcl-3, little is known about its cell-specific functions or mechanisms. Here we demonstrate a T-cell-intrinsic function of Bcl-3 in autoimmunity. Bcl-3-deficient T cells failed to induce disease in T cell transfer-induced colitis and experimental autoimmune encephalomyelitis. The protection against disease correlated with a decrease in Th1 cells that produced the cytokines IFN-gamma and GM-CSF and an increase in Th17 cells. Although differentiation into Th1 cells was not impaired in the absence of Bcl-3, differentiated Th1 cells converted to less-pathogenic Th17-like cells, in part via mechanisms involving expression of the RORgammat transcription factor. Thus, Bcl-3 constrained Th1 cell plasticity and promoted pathogenicity by blocking conversion to Th17-like cells, revealing a unique type of regulation that shapes adaptive immunity.
-
Berger, H., et al (2013). "SOCS3 transactivation by PPARgamma prevents IL-17-driven cancer growth" Cancer Res 73(12): 3578-3590.
PubMed
Activation of the transcription factor PPARgamma by the n-3 fatty acid docosahexaenoic acid (DHA) is implicated in controlling proinflammatory cytokine secretion, but the intracellular signaling pathways engaged by PPARgamma are incompletely characterized. Here, we identify the adapter-encoding gene SOCS3 as a critical transcriptional target of PPARgamma. SOCS3 promoter binding and gene transactivation by PPARgamma was associated with a repression in differentiation of proinflammatory T-helper (TH)17 cells. Accordingly, TH17 cells induced in vitro displayed increased SOCS3 expression and diminished capacity to produce interleukin (IL)-17 following activation of PPARgamma by DHA. Furthermore, naive CD4 T cells derived from mice fed a DHA-enriched diet displayed less capability to differentiate into TH17 cells. In two different mouse models of cancer, DHA prevented tumor outgrowth and angiogenesis in an IL-17-dependent manner. Altogether, our results uncover a novel molecular pathway by which PPARgamma-induced SOCS3 expression prevents IL-17-mediated cancer growth.
-
Bertin, S., et al (2014). "The ion channel TRPV1 regulates the activation and proinflammatory properties of CD4(+) T cells" Nat Immunol 15(11): 1055-1063.
PubMed
TRPV1 is a Ca(2+)-permeable channel studied mostly as a pain receptor in sensory neurons. However, its role in other cell types is poorly understood. Here we found that TRPV1 was functionally expressed in CD4(+) T cells, where it acted as a non-store-operated Ca(2+) channel and contributed to T cell antigen receptor (TCR)-induced Ca(2+) influx, TCR signaling and T cell activation. In models of T cell-mediated colitis, TRPV1 promoted colitogenic T cell responses and intestinal inflammation. Furthermore, genetic and pharmacological inhibition of TRPV1 in human CD4(+) T cells recapitulated the phenotype of mouse Trpv1(-/-) CD4(+) T cells. Our findings suggest that inhibition of TRPV1 could represent a new therapeutic strategy for restraining proinflammatory T cell responses.
Product Citations
-
IFITM1 is required for epithelial mesenchymal transition in airway remodeling of allergic asthma.
In World Allergy Organ J on 1 March 2026 by Zhu, M., Weng, X., et al.
PubMed
Interferon-induced transmembrane protein 1 (IFITM1) restricts virus infection. IFITM proteins are involved in Th2 cell differentiation in allergic asthma. The epithelial‒mesenchymal transition (EMT) regulates allergic airway remodeling. We sought to explore the functional contributions and underlying mechanisms of IFITM1 in the EMT associated with allergic asthma.
-
CEBPB Expression in Tumor Cells Drives Immune Evasion in Colorectal Cancer via CTLA4 Up-regulation in T Cells.
In Cancer Commun (Lond) on 26 February 2026 by Yun, H. J., Park, C. H., et al.
PubMed
Background: Immune checkpoint inhibitors are ineffective in the majority of colorectal cancers (CRCs) that are microsatellite stable. However, the underlying reasons for their unresponsiveness and mechanisms of immune evasion are poorly understood. In the present study, we aimed to elucidate the mechanisms underlying the immune evasion driven by CRC cells. Methods: We performed single-cell RNA sequencing of tumor tissues from 30 CRC patients and syngeneic mice implanted with transformation-related protein 53 (Trp53) knockout CT26 cells. Gene expression and correlations of individual tumor microenvironment (TME) components were analyzed, and their functional significance was investigated using syngeneic mouse models and cell line co-culture experiments. Results: CCAAT enhancer-binding protein beta (CEBPB) expression was increased in tumor protein 53 (TP53)-mutated CRCs. We confirmed that wild-type TP53 negatively regulated CEBPB expression in CRC cell lines. CEBPB expression was associated with decreased intratumoral T cell infiltration and negatively impacted survival in CRC patients. In the intercellular correlation analysis of gene expression, tumor epithelial cell CEBPB expression was significantly correlated with cytotoxic T-lymphocyte associated protein 4 (CTLA4) expression in T cells, especially in regulatory and exhausted T cells. Cebpb overexpression promoted tumor growth in the immunocompetent syngeneic mouse models, which was accompanied by increased CTLA-4 expression in tumor-infiltrating CD4+ T cells. In vitro co-culture experiments also showed that tumor cell CEBPB overexpression increased CTLA4 in T cells. Conclusions: Tumor cell CEBPB expression, up-regulated by TP53 mutation, can increase CTLA4 expression in T cells and negatively affect patient outcomes. These findings suggested a central role of tumor cell CEBPB in shaping an immunosuppressive TME.
-
Escherichia coli promotes colorectal cancer metastasis by maintaining enhancer-promoter loops through releasing neutrophil extracellular traps.
In Nat Commun on 3 February 2026 by Pan, B., Yao, Y., et al.
PubMed
The involvement of intestinal microbiota in the process of neutrophil-mediated colorectal cancer liver metastasis (CRCLM) is not yet fully understood. Here, we show that Escherichia coli (E. coli) is prevalent in CRC tissues with LM using 2bRAD-M-Seq and is linked to the release of neutrophil extracellular traps (NETs). Utilizing multi-omics and molecular techniques, we establish that E. coli recruits RIPK2, which promotes the binding of HNRNPK to the Atf3/Relb promoters in neutrophils, thereby enhancing their transcription. This process results in the upregulation of Ncf4, which triggers p-MLKL-mediated NET formation. NETs, in turn, increase the expression of TRPC1 and NFATC3 in CRC cells, promoting the calcium-dependent assembly of the STAT3/S100A8/9 heterotrimer. This trimer stabilizes STAT3-enhancer-promoter loops (EPLs), thereby reinforcing the Tns1 transcription and facilitating CRCLM. Our findings elucidate the mechanism by which E. coli-induced NETs promote CRCLM through epigenetic modifications, offering an insight into the role of EPLs in immune regulation and tumor progression.
-
Reprogramming the melanoma tumor immune microenvironment via combinatorial signal 2/3 gene delivery.
In J Immunother Cancer on 27 January 2026 by Luly, K. M., Zhou, X. M. M., et al.
PubMed
An adaptive immune response to cancer requires three main signals: antigen presentation and recognition ("signal 1"), costimulation ("signal 2"), and secreted immunostimulatory cytokines ("signal 3"). Expression of these signals in tumors via non-viral gene delivery represents a promising strategy to reprogram the tumor microenvironment (TME) and prime antitumor immunity.