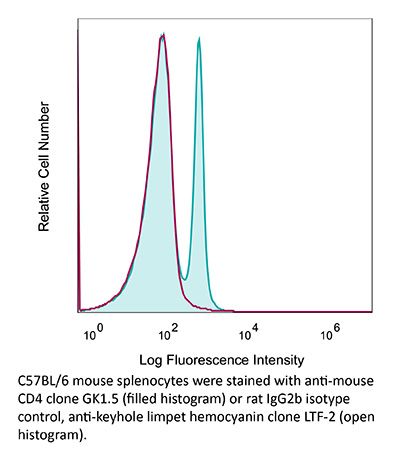
InVivoMAb anti-mouse CD4
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse CTL clone V4 |
| Reported Applications |
in vivo CD4+ T cell depletion Flow cytometry Western blot |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107636 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Hervieu, A., et al (2013). "Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth" J Invest Dermatol 133(2): 499-508.
PubMed
Dacarbazine (DTIC) is a cytotoxic drug widely used for melanoma treatment. However, the putative contribution of anticancer immune responses in the efficacy of DTIC has not been evaluated. By testing how DTIC affects host immune responses to cancer in a mouse model of melanoma, we unexpectedly found that both natural killer (NK) and CD8(+) T cells were indispensable for DTIC therapeutic effect. Although DTIC did not directly affect immune cells, it triggered the upregulation of NKG2D ligands on tumor cells, leading to NK cell activation and IFNgamma secretion in mice and humans. NK cell-derived IFNgamma subsequently favored upregulation of major histocompatibility complex class I molecules on tumor cells, rendering them sensitive to cytotoxic CD8(+) T cells. Accordingly, DTIC markedly enhanced cytotoxic T lymphocyte antigen 4 inhibition efficacy in vivo in an NK-dependent manner. These results underscore the immunogenic properties of DTIC and provide a rationale to combine DTIC with immunotherapeutic agents that relieve immunosuppression in vivo.
-
Hervieu, A., et al (2013). "Dacarbazine-mediated upregulation of NKG2D ligands on tumor cells activates NK and CD8 T cells and restrains melanoma growth" J Invest Dermatol 133(2): 499-508.
PubMed
Dacarbazine (DTIC) is a cytotoxic drug widely used for melanoma treatment. However, the putative contribution of anticancer immune responses in the efficacy of DTIC has not been evaluated. By testing how DTIC affects host immune responses to cancer in a mouse model of melanoma, we unexpectedly found that both natural killer (NK) and CD8(+) T cells were indispensable for DTIC therapeutic effect. Although DTIC did not directly affect immune cells, it triggered the upregulation of NKG2D ligands on tumor cells, leading to NK cell activation and IFNgamma secretion in mice and humans. NK cell-derived IFNgamma subsequently favored upregulation of major histocompatibility complex class I molecules on tumor cells, rendering them sensitive to cytotoxic CD8(+) T cells. Accordingly, DTIC markedly enhanced cytotoxic T lymphocyte antigen 4 inhibition efficacy in vivo in an NK-dependent manner. These results underscore the immunogenic properties of DTIC and provide a rationale to combine DTIC with immunotherapeutic agents that relieve immunosuppression in vivo.
-
Dai, M., et al (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257.
PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
-
Dai, M., et al (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257.
PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
Product Citations
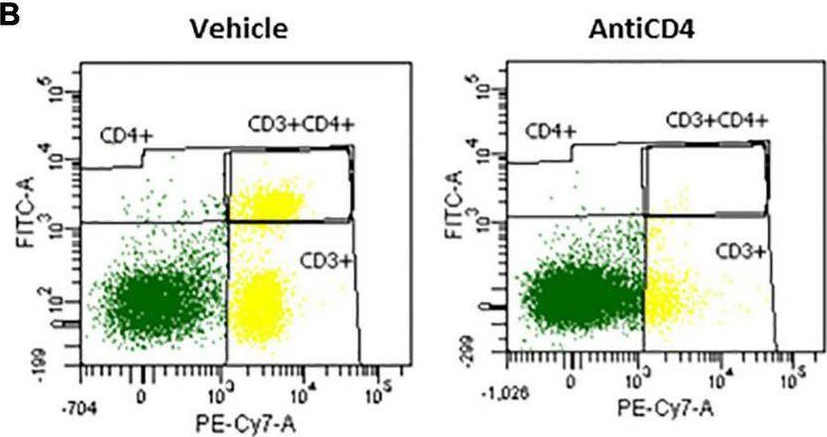
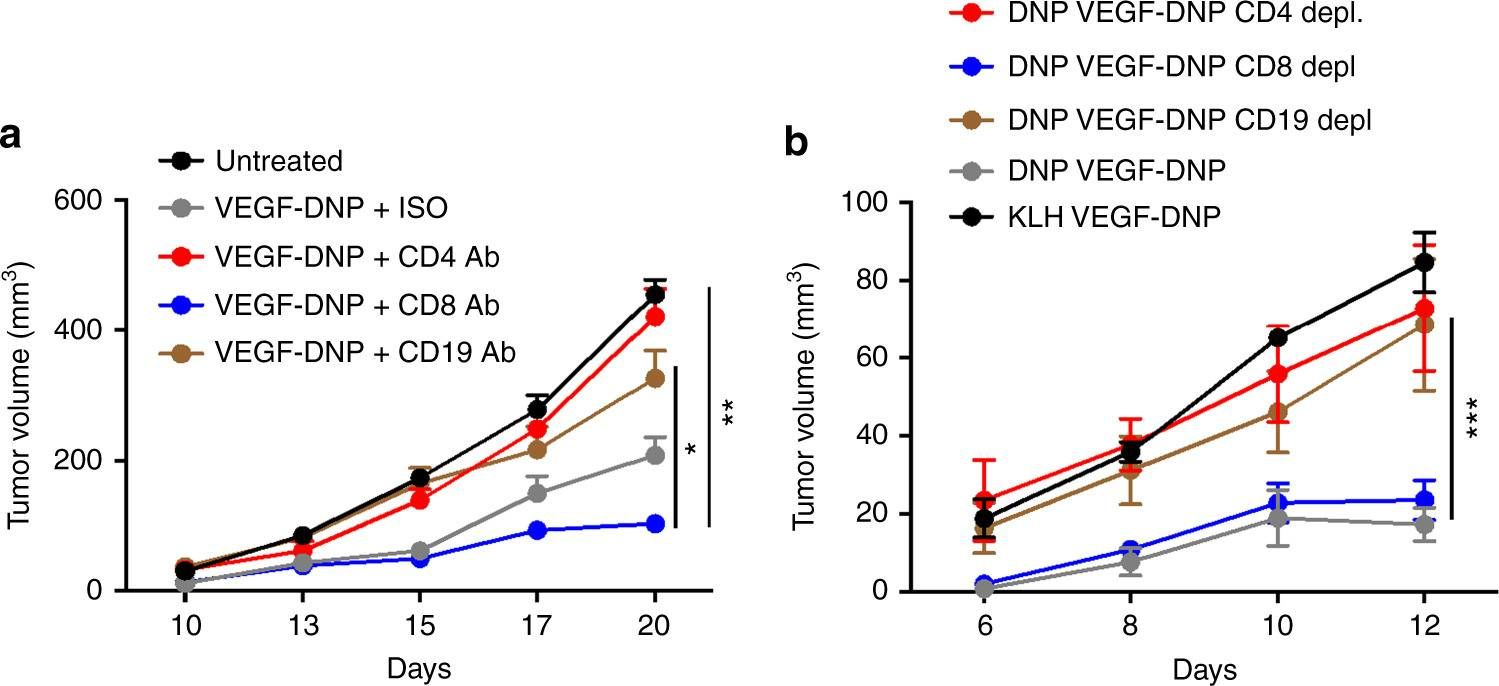
-
Microbiota-induced T cell plasticity enables immune-mediated tumour control.
In Nature on 1 March 2026 by Najar, T. A., Hao, Y., et al.
PubMed
Therapies that harness the immune system to target and eliminate tumour cells have revolutionized cancer care. Immune checkpoint blockade (ICB), which boosts the anti-tumour immune response by inhibiting negative regulators of T cell activation1-3, is remarkably successful in a subset of cancer patients. Yet a significant proportion do not respond to treatment, emphasizing the need to understand factors influencing the therapeutic efficacy of ICB4-9. The gut microbiota, consisting of trillions of microorganisms residing in the gastrointestinal tract, has emerged as a critical determinant of immune function and response to cancer immunotherapy, with several studies demonstrating association of microbiota composition with clinical response10-16. However, a mechanistic understanding of how gut commensal bacteria influence the efficacy of ICB remains elusive. Here we use a gut commensal microorganism, segmented filamentous bacteria (SFB), which induces an antigen-specific T helper 17 (TH17) cell effector program in the small intestine lamina propria (SILP)17, to investigate how colonization with this microbe affects the efficacy of ICB in restraining distal growth of tumours sharing antigen with SFB. We find that anti-programmed cell death protein 1 (PD-1) treatment effectively inhibits the growth of implanted SFB antigen-expressing melanoma only if mice are colonized with SFB. Through T cell receptor (TCR) clonal lineage tracing, fate mapping and peptide-major histocompatability complex (MHC) tetramer staining, we identify tumour-associated SFB-specific T helper 1 (TH1)-like cells derived from the homeostatic TH17 cells induced by SFB colonization in the SILP. These gut-educated ex-TH17 cells produce high levels of the pro-inflammatory cytokines interferon (IFN)-γ and tumour necrosis factor (TNF) within the tumour microenvironment (TME), enhancing antigen presentation and promoting recruitment, expansion and effector functions of CD8+ tumour-infiltrating cytotoxic lymphocytes and thereby enabling anti-PD-1-mediated tumour control. Conditional ablation of SFB-induced IL-17A+CD4+ T cells, precursors of tumour-associated TH1-like cells, abolishes anti-PD-1-mediated tumour control and markedly impairs tumour-specific CD8+ T cell recruitment and effector function within the TME. Our data, as a proof of principle, define a cellular pathway by which a single, defined intestinal commensal imprints T cell plasticity that potentiates PD-1 blockade, and indicate targeted modulation of the microbiota as a strategy to broaden ICB efficacy.
-
HRS Degradation-Induced Nicotinamide Deficiency in Placental Extracellular Vesicles Triggers Preeclampsia by Disrupting Maternal-Fetal Immune Homeostasis.
In Adv Sci (Weinh) on 1 February 2026 by Fei, H., Lin, Y., et al.
PubMed
Preeclampsia (PE) is closely associated with alterations in placental extracellular vesicles (pEVs), but the mechanisms and their role in PE pathogenesis remain unclear. This study reveals that nicotinamide (NAM) levels in PE-derived pEVs (PE-EVs) are lower than in pEVs from normal pregnancies, correlating with disease severity. Functionally, NAM in pEVs inhibits Th1 differentiation via SIRT1 suppression and Th17 differentiation via macrophages. NAM-deficient pEVs exhibit reduced capacity to inhibit Th1 and Th17 cell differentiation both in vitro and in vivo, leading to PE-like symptoms. NAM is enriched in pEVs compared to placental villous tissue and maternal serum. The lower NAM in PE-EVs is due to decreased hepatocyte growth factor-regulated tyrosine kinase substrate (HRS) expression in trophoblasts, which loads NAM into the cargo of multivesicular bodies (MVBs) via binding to the tryptophan-115 residue of HRS. Furthermore, the reduction of HRS in PE trophoblasts results from ubiquitination and degradation by elevated HSP27. Collectively, these findings indicate that elevated HSP27 in PE trophoblasts leads to the degradation of HRS, a reduction in pEV NAM levels, and diminished Th1 and Th17 inhibitory effects, thereby contributing to the development of PE.
-
SLC2A1+ tumour-associated macrophages spatially control CD8+ T cell function and drive resistance to immunotherapy in non-small-cell lung cancer.
In Nat Cell Biol on 1 February 2026 by Wang, L., Chu, H., et al.
PubMed
Tumour-associated macrophages (TAMs) contribute to immune checkpoint blockade resistance, but their impact on intratumoural CD8⁺ T cell distribution remains unclear. Here we show that the expression of the glucose transporter SLC2A1 is spatially negatively correlated with CD8⁺ T cell distribution in both non-small-cell lung cancer (NSCLC) biopsies and murine tumour models. Tumour cell-specific Slc2a1 knockdown fails to reproduce the therapeutic benefit of SLC2A1 inhibition, whereas TAM-specific deletion of Slc2a1 suppresses tumour growth by enhancing the spatial homogeneity and effector function of intratumoural CD8⁺ T cells, thereby improving αPD-L1 efficacy. Spatial profiling of NSCLC specimens further revealed that SLC2A1⁺ TAM-enriched regions exhibit reduced CD8⁺ T cell density, and spatial proximity between these populations predicts resistance to αPD-(L)1 therapy. These findings identify SLC2A1⁺ TAMs as drivers of spatial CD8⁺ T cell exclusion and highlight TAM-specific SLC2A1 as a therapeutic target to overcome immune checkpoint blockade resistance in NSCLC.
-
Cathepsin-D-mediated MHC class I degradation contributes to immune evasion in colorectal cancer.
In Cell Rep Med on 20 January 2026 by Zhan, W., Fu, Y., et al.
PubMed
Microsatellite stable (MSS) colorectal cancer (CRC) is often considered a "cold" tumor with limited response to programmed death-1 (PD-1) antibody monotherapy. The mechanisms underlying its intrinsic resistance to immunotherapy remain unclear. Here, we show that cathepsin D (CTSD) is highly expressed in MSS CRC and contributes significantly to immunotherapy resistance. Mechanistically, CTSD, acting as a protease, interacts with the α2 domain of the major histocompatibility complex (MHC) class I via the light chain of its catalytic domain, promoting MHC class I degradation through lysosomal pathways and impairing its recycling to the cell surface. This mechanism shields tumor cells from cytotoxic T-cell-mediated killing and facilitates immune evasion. Notably, genetic deletion or pharmacological inhibition of CTSD using pepstatin A prevents immune escape and enhances anti-PD-1 efficacy. These findings identify CTSD as a key mediator of immune evasion in MSS CRC and support the development of a combination therapy comprising CTSD inhibition and anti-PD-1 immunotherapy.