InVivoMAb anti-mouse Ly6G/Ly6C (Gr-1)
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
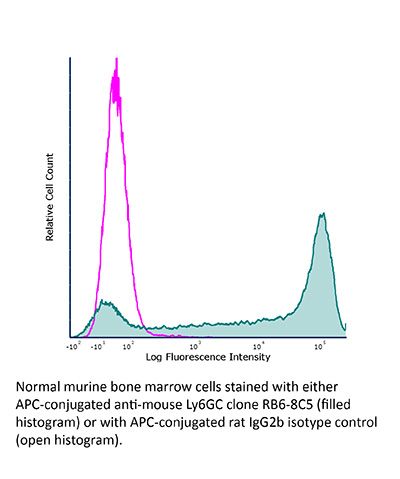
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse granulocytes |
| Reported Applications |
in vivo depletion of Gr-1+ myeloid cells Flow cytometry Immunohistochemistry (paraffin) Immunohistochemistry (frozen) |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10312146 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
in vivo CD16.2 blockade
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Schulze, F. S., et al (2014). "Fcgamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid" Am J Pathol 184(8): 2185-2196.
PubMed
Bullous pemphigoid (BP) and epidermolysis bullosa acquisita are subepidermal autoimmune blistering diseases mediated by autoantibodies against type XVII collagen (Col17) and Col7, respectively. For blister formation, Fc-mediated events, such as infiltration of inflammatory cells in the skin, complement activation, and release of proteases at the dermal-epidermal junction, are essential. Although in the neonatal passive transfer mouse model of BP, tissue destruction is mediated by Fcgamma receptors (FcgammaRs) I and III, the passive transfer model of epidermolysis bullosa acquisita completely depends on FcgammaRIV. To clarify this discrepancy, we developed a novel experimental model for BP using adult mice. Lesion formation was Fc mediated because gamma-chain-deficient mice and mice treated with anti-Col17 IgG, depleted from its sugar moiety at the Fc portion, were resistant to disease induction. By the use of various FcgammaR-deficient mouse strains, tissue destruction was shown to be mediated by FcgammaRIV, FcgammaRIII, and FcgammaRIIB, whereas FcgammaRI was not essential. Furthermore, anti-inflammatory mediators in already clinically diseased mice can be explored in the novel BP model, because the pharmacological inhibition of FcgammaRIV and depletion of granulocytes abolished skin blisters. Herein, we extended our knowledge about the importance of FcgammaRs in experimental BP and established a novel BP mouse model suitable to study disease development over a longer time period and explore novel treatment strategies in a quasi-therapeutic setting.
in vivo CD16.2 blockade
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Schulze, F. S., et al (2014). "Fcgamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid" Am J Pathol 184(8): 2185-2196.
PubMed
Bullous pemphigoid (BP) and epidermolysis bullosa acquisita are subepidermal autoimmune blistering diseases mediated by autoantibodies against type XVII collagen (Col17) and Col7, respectively. For blister formation, Fc-mediated events, such as infiltration of inflammatory cells in the skin, complement activation, and release of proteases at the dermal-epidermal junction, are essential. Although in the neonatal passive transfer mouse model of BP, tissue destruction is mediated by Fcgamma receptors (FcgammaRs) I and III, the passive transfer model of epidermolysis bullosa acquisita completely depends on FcgammaRIV. To clarify this discrepancy, we developed a novel experimental model for BP using adult mice. Lesion formation was Fc mediated because gamma-chain-deficient mice and mice treated with anti-Col17 IgG, depleted from its sugar moiety at the Fc portion, were resistant to disease induction. By the use of various FcgammaR-deficient mouse strains, tissue destruction was shown to be mediated by FcgammaRIV, FcgammaRIII, and FcgammaRIIB, whereas FcgammaRI was not essential. Furthermore, anti-inflammatory mediators in already clinically diseased mice can be explored in the novel BP model, because the pharmacological inhibition of FcgammaRIV and depletion of granulocytes abolished skin blisters. Herein, we extended our knowledge about the importance of FcgammaRs in experimental BP and established a novel BP mouse model suitable to study disease development over a longer time period and explore novel treatment strategies in a quasi-therapeutic setting.
in vivo CD16.2 blockade
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Schulze, F. S., et al (2014). "Fcgamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid" Am J Pathol 184(8): 2185-2196.
PubMed
Bullous pemphigoid (BP) and epidermolysis bullosa acquisita are subepidermal autoimmune blistering diseases mediated by autoantibodies against type XVII collagen (Col17) and Col7, respectively. For blister formation, Fc-mediated events, such as infiltration of inflammatory cells in the skin, complement activation, and release of proteases at the dermal-epidermal junction, are essential. Although in the neonatal passive transfer mouse model of BP, tissue destruction is mediated by Fcgamma receptors (FcgammaRs) I and III, the passive transfer model of epidermolysis bullosa acquisita completely depends on FcgammaRIV. To clarify this discrepancy, we developed a novel experimental model for BP using adult mice. Lesion formation was Fc mediated because gamma-chain-deficient mice and mice treated with anti-Col17 IgG, depleted from its sugar moiety at the Fc portion, were resistant to disease induction. By the use of various FcgammaR-deficient mouse strains, tissue destruction was shown to be mediated by FcgammaRIV, FcgammaRIII, and FcgammaRIIB, whereas FcgammaRI was not essential. Furthermore, anti-inflammatory mediators in already clinically diseased mice can be explored in the novel BP model, because the pharmacological inhibition of FcgammaRIV and depletion of granulocytes abolished skin blisters. Herein, we extended our knowledge about the importance of FcgammaRs in experimental BP and established a novel BP mouse model suitable to study disease development over a longer time period and explore novel treatment strategies in a quasi-therapeutic setting.
Immunohistochemistry (paraffin)
Li, M., et al (2006). "Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis" Proc Natl Acad Sci U S A 103(31): 11736-11741.
PubMed
We have demonstrated that cytokine thymic stromal lymphopoietin (TSLP), whose expression is rapidly induced upon keratinocyte-selective ablation of retinoid X receptors (RXRs) -alpha and -beta in the mouse (RXRalphabeta(ep-/-) mice), plays a key role in initiating a skin and systemic atopic dermatitis-like phenotype. We show here that topical application of the physiologically active ligand [1alpha,25-(OH)(2)D(3); calcitriol] of the vitamin D receptor, or of its low-calcemic analog MC903 (calcipotriol; Dovonex), induces TSLP expression in epidermal keratinocytes, which results in an atopic dermatitis-like syndrome mimicking that seen in RXRalphabeta(ep-/-) mutants and transgenic mice overexpressing TSLP in keratinocytes. Furthermore, topical application of retinoic acid receptor RARgamma-selective agonist BMS961 also induces TSLP expression either on its own or synergistically with 1alpha,25-(OH)(2)D(3). Our data demonstrate that RXR/vitamin D receptor and RXR/retinoic acid receptor-gamma heterodimers and their ligands cell-autonomously control the expression of TSLP in epidermal keratinocytes of the mouse. We propose molecular mechanisms through which vitamin D3 and retinoic acid signalings could be involved in the pathogenesis of atopic diseases.
in vivo IL-17A neutralization
in vivo depletion of Gr-1+ myeloid cells
in vivo ILC depletion
Ermann, J., et al (2014). "Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice" Proc Natl Acad Sci U S A 111(25): E2559-2566.
PubMed
T-bet(-/-).Rag2(-/-) (TRUC) mice spontaneously develop microbiota-driven, TNF-mediated large bowel inflammation that resembles human ulcerative colitis. We show here that IL-23 and IL-1-dependent secretion of IL-17A by innate lymphoid cells (ILCs; defined as CD45(+)lin(-)Thy1(hi)NKp46(-)) is a second critical pathway in this model. Using an in vitro coculture system of bone marrow-derived dendritic cells (DCs) and freshly isolated FACS-purified ILCs, we demonstrate that IL-23 and IL-1 secreted by DCs in response to microbial stimulation work together to induce IL-17A production by ILCs. TNF is not required for IL-17A secretion by ILCs in vitro but synergizes with IL-17A to induce the expression of neutrophil-attracting chemokines. Upstream, activation of the IL-23/IL-17A axis is regulated by nucleotide-binding oligomerization domain containing (Nod)/receptor-interacting serine-threonine kinase 2 (Ripk2) signals in DCs. Genetic ablation of the Nod/Ripk2 signaling pathway protects TRUC mice from developing colitis without affecting the colitogenicity of the intestinal microbiota. Our data provide insight into the complex network of interactions between IL-17A-secreting ILCs and other components of the innate immune system in the development of colitis.
Immunohistochemistry (frozen)
Brown, C. R., et al (2004). "Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental lyme arthritis via the recruitment of Gr-1- polymorphonuclear leukocyte-like cells" Infect Immun 72(9): 4956-49
PubMed
Recently, we demonstrated that blocking the entry of neutrophils into Borrelia burgdorferi-infected joints in mice deficient in the chemokine receptor CXCR2 prevented the development of experimental Lyme arthritis. Neutrophils were marginalized in blood vessels at the site of infection but could not enter the joint tissue. In the present study, we treated both genetically arthritis-resistant DBA/2J (DBA) and arthritis-susceptible C3H/HeJ (C3H) mice with the neutrophil-depleting monoclonal antibody RB6-8C5 (RB6) to determine the effect on arthritis development. Surprisingly, both DBA and C3H mice treated with RB6 developed arthritis at 1 week postinfection, approximately 1 week earlier than the control-treated C3H mice. The early development of arthritis in the RB6-treated mice was accompanied by an influx into the joints of cells with ring-shaped polymorphonuclear leukocyte (PMN) cell morphology that were negative for the Gr-1 neutrophil maturation marker. RB6 treatment of mice also resulted in increased numbers of B. burgdorferi cells in the joints at 7 days postinfection and earlier expression of the chemokines KC and monocyte chemoattractant protein 1 in the joints compared to control-treated animals. Together, these results suggest that recruitment of neutrophils or PMN-like cells into an infected joint is a key requirement for Lyme arthritis development and that altered recruitment of these cells into the joints of arthritis-resistant mice can exacerbate the development of pathology.
in vivo IL-17A neutralization
in vivo depletion of Gr-1+ myeloid cells
in vivo ILC depletion
Ermann, J., et al (2014). "Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice" Proc Natl Acad Sci U S A 111(25): E2559-2566.
PubMed
T-bet(-/-).Rag2(-/-) (TRUC) mice spontaneously develop microbiota-driven, TNF-mediated large bowel inflammation that resembles human ulcerative colitis. We show here that IL-23 and IL-1-dependent secretion of IL-17A by innate lymphoid cells (ILCs; defined as CD45(+)lin(-)Thy1(hi)NKp46(-)) is a second critical pathway in this model. Using an in vitro coculture system of bone marrow-derived dendritic cells (DCs) and freshly isolated FACS-purified ILCs, we demonstrate that IL-23 and IL-1 secreted by DCs in response to microbial stimulation work together to induce IL-17A production by ILCs. TNF is not required for IL-17A secretion by ILCs in vitro but synergizes with IL-17A to induce the expression of neutrophil-attracting chemokines. Upstream, activation of the IL-23/IL-17A axis is regulated by nucleotide-binding oligomerization domain containing (Nod)/receptor-interacting serine-threonine kinase 2 (Ripk2) signals in DCs. Genetic ablation of the Nod/Ripk2 signaling pathway protects TRUC mice from developing colitis without affecting the colitogenicity of the intestinal microbiota. Our data provide insight into the complex network of interactions between IL-17A-secreting ILCs and other components of the innate immune system in the development of colitis.
in vivo IL-17A neutralization
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
in vivo TNFα neutralization
Flow Cytometry
in vivo IL-6 neutralization
in vivo IL-1β neutralization
Khmaladze, I., et al (2014). "Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice" Proc Natl Acad Sci U S A 111(35): E3669-3678.
PubMed
Psoriasis (Ps) and psoriasis arthritis (PsA) are poorly understood common diseases, induced by unknown environmental factors, affecting skin and articular joints. A single i.p. exposure to mannan from Saccharomyces cerevisiae induced an acute inflammation in inbred mouse strains resembling human Ps and PsA-like disease, whereas multiple injections induced a relapsing disease. Exacerbation of disease severity was observed in mice deficient for generation of reactive oxygen species (ROS). Interestingly, restoration of ROS production, specifically in macrophages, ameliorated both skin and joint disease. Neutralization of IL-17A, mainly produced by gammadelta T cells, completely blocked disease symptoms. Furthermore, mice depleted of granulocytes were resistant to disease development. In contrast, certain acute inflammatory mediators (C5, Fcgamma receptor III, mast cells, and histamine) and adaptive immune players (alphabeta T and B cells) were redundant in disease induction. Hence, we propose that mannan-induced activation of macrophages leads to TNF-alpha secretion and stimulation of local gammadelta T cells secreting IL-17A. The combined action of activated macrophages and IL-17A produced in situ drives neutrophil infiltration in the epidermis and dermis of the skin, leading to disease manifestations. Thus, our finding suggests a new mechanism triggered by exposure to exogenous microbial components, such as mannan, that can induce and exacerbate Ps and PsA.
in vivo depletion of Gr-1+ myeloid cells
Bansal, S., et al (2018). "IL-1 Signaling Prevents Alveolar Macrophage Depletion during Influenza and Streptococcus pneumoniae Coinfection" J Immunol 200(4): 1425-1433.
PubMed
Influenza and bacterial coinfection is a significant cause of hospitalization and death in humans during influenza epidemics and pandemics. However, the fundamental protective and pathogenic mechanisms involved in this complex virus-host-bacterium interaction remain incompletely understood. In this study, we have developed mild to lethal influenza and Streptococcus pneumoniae coinfection models for comparative analyses of disease pathogenesis. Specifically, wild-type and IL-1R type 1-deficient (Il1r1(-/-) ) mice were infected with influenza virus and then superchallenged with noninvasive S. pneumoniae serotype 14 (Spn14) or S. pneumoniae serotype 19A (Spn19A). The coinfections were followed by comparative analyses of inflammatory responses and animal protection. We found that resident alveolar macrophages are efficient in the clearance of both pneumococcal serotypes in the absence of influenza infection; in contrast, they are essential for airway control of Spn14 infection but not Spn19A infection. In agreement, TNF-alpha and neutrophils play a compensatory protective role in secondary bacterial infection associated with Spn19A; however, the essential requirement for alveolar macrophage-mediated clearance significantly enhances the virulence of Spn14 during postinfluenza pneumococcal infection. Furthermore, we show that, although IL-1 signaling is not required for host defense against pneumococcal infection alone, it is essential for sustaining antibacterial immunity during postinfluenza pneumococcal infection, as evidenced by significantly aggravated bacterial burden and animal mortality in Il1r1(-/-) mice. Mechanistically, we show that through preventing alveolar macrophage depletion, inflammatory cytokine IL-1 signaling is critically involved in host resistance to influenza and pneumococcal coinfection.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
in vitro TGFβ neutralization
Bodogai, M., et al (2015). "Immunosuppressive and Prometastatic Functions of Myeloid-Derived Suppressive Cells Rely upon Education from Tumor-Associated B Cells" Cancer Res 75(17): 3456-3465.
PubMed
Myeloid-derived suppressive cells (MDSC) have been reported to promote metastasis, but the loss of cancer-induced B cells/B regulatory cells (tBreg) can block metastasis despite MDSC expansion in cancer. Here, using multiple murine tumor models and human MDSC, we show that MDSC populations that expand in cancer have only partially primed regulatory function and limited prometastatic activity unless they are fully educated by tBregs. Cancer-induced tBregs directly activate the regulatory function of both the monocyte and granulocyte subpopulations of MDSC, relying, in part, on TgfbetaR1/TgfbetaR2 signaling. MDSC fully educated in this manner exhibit an increased production of reactive oxygen species and NO and more efficiently suppress CD4(+) and CD8(+) T cells, thereby promoting tumor growth and metastasis. Thus, loss of tBregs or TgfbetaR deficiency in MDSC is sufficient to disable their suppressive function and to block metastasis. Overall, our data indicate that cancer-induced B cells/B regulatory cells are important regulators of the immunosuppressive and prometastatic functions of MDSC.
in vivo depletion of Gr-1+ myeloid cells
Dahlgren, M. W., et al (2015). "T follicular helper, but not Th1, cell differentiation in the absence of conventional dendritic cells" J Immunol 194(11): 5187-5199.
PubMed
Development of long-lived humoral immunity is dependent on CXCR5-expressing T follicular helper (Tfh) cells, which develop concomitantly to effector Th cells that support cellular immunity. Conventional dendritic cells (cDCs) are critical APCs for initial priming of naive CD4(+) T cells but, importantly, also provide accessory signals that govern effector Th cell commitment. To define the accessory role of cDCs during the concurrent development of Tfh and effector Th1 cells, we performed high-dose Ag immunization in conjunction with the Th1-biased adjuvant polyinosinic:polycytidylic acid (pI:C). In the absence of cDCs, pI:C failed to induce Th1 cell commitment and IgG2c production. However, cDC depletion did not impair Tfh cell differentiation or germinal center formation, and long-lived IgG1 responses of unaltered affinity developed in mice lacking cDCs at the time point for immunization. Thus, cDCs are required for the pI:C-driven Th1 cell fate commitment but have no crucial accessory function in relation to Tfh cell differentiation.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Wang, H., et al (2015). "P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy" Cell Res 25(6): 674-690.
PubMed
Septic encephalopathy (SE) is a critical factor determining sepsis mortality. Vascular inflammation is known to be involved in SE, but the molecular events that lead to the development of encephalopathy remain unclear. Using time-lapse in vivo two-photon laser scanning microscopy, we provide the first direct evidence that cecal ligation and puncture in septic mice induces microglial trafficking to sites adjacent to leukocyte adhesion on inflamed cerebral microvessels. Our data further demonstrate that septic injury increased the chemokine CXCL1 level in brain endothelial cells by activating endothelial P2RX7 and eventually enhanced the binding of Mac-1 (CD11b/CD18)-expressing leukocytes to endothelial ICAM-1. In turn, leukocyte adhesion upregulated endothelial CX3CL1, thereby triggering microglia trafficking to the injured site. The sepsis-induced increase in endothelial CX3CL1 was abolished in CD18 hypomorphic mutant mice. Inhibition of the P2RX7 pathway not only decreased endothelial ICAM-1 expression and leukocyte adhesion but also prevented microglia overactivation, reduced brain injury, and consequently doubled the early survival of septic mice. These results demonstrate the role of the P2RX7 pathway in linking neurovascular inflammation to brain damage in vivo and provide a rationale for targeting endothelial P2RX7 for neurovascular protection during SE.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Bryant, J., et al (2014). "Preemptive donor apoptotic cell infusions induce IFN-gamma-producing myeloid-derived suppressor cells for cardiac allograft protection" J Immunol 192(12): 6092-6101.
PubMed
We have previously shown that preemptive infusion of apoptotic donor splenocytes treated with the chemical cross-linker ethylcarbodiimide (ECDI-SPs) induces long-term allograft survival in full MHC-mismatched models of allogeneic islet and cardiac transplantation. The role of myeloid-derived suppressor cells (MDSCs) in the graft protection provided by ECDI-SPs is unclear. In this study, we demonstrate that infusions of ECDI-SPs increase two populations of CD11b(+) cells in the spleen that phenotypically resemble monocytic-like (CD11b(+)Ly6C(high)) and granulocytic-like (CD11b(+)Gr1(high)) MDSCs. Both populations suppress T cell proliferation in vitro and traffic to the cardiac allografts in vivo to mediate their protection via inhibition of local CD8 T cell accumulation and potentially also via induction and homing of regulatory T cells. Importantly, repeated treatments with ECDI-SPs induce the CD11b(+)Gr1(high) cells to produce a high level of IFN-gamma and to exhibit an enhanced responsiveness to IFN-gamma by expressing higher levels of downstream effector molecules ido and nos2. Consequently, neutralization of IFN-gamma completely abolishes the suppressive capacity of this population. We conclude that donor ECDI-SPs induce the expansion of two populations of MDSCs important for allograft protection mediated in part by intrinsic IFN-gamma-dependent mechanisms. This form of preemptive donor apoptotic cell infusions has significant potential for the therapeutic manipulation of MDSCs for transplant tolerance induction.
in vivo depletion of Gr-1+ myeloid cells
in vivo CTLA-4 neutralization
in vivo induction TRAIL-mediated apoptosis
in vitro induction TRAIL-mediated apoptosis
Condamine, T., et al (2014). "ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis" J Clin Invest 124(6): 2626-2639.
PubMed
Myeloid-derived suppressor cells (MDSCs) dampen the immune response thorough inhibition of T cell activation and proliferation and often are expanded in pathological conditions. Here, we studied the fate of MDSCs in cancer. Unexpectedly, MDSCs had lower viability and a shorter half-life in tumor-bearing mice compared with neutrophils and monocytes. The reduction of MDSC viability was due to increased apoptosis, which was mediated by increased expression of TNF-related apoptosis-induced ligand receptors (TRAIL-Rs) in these cells. Targeting TRAIL-Rs in naive mice did not affect myeloid cell populations, but it dramatically reduced the presence of MDSCs and improved immune responses in tumor-bearing mice. Treatment of myeloid cells with proinflammatory cytokines did not affect TRAIL-R expression; however, induction of ER stress in myeloid cells recapitulated changes in TRAIL-R expression observed in tumor-bearing hosts. The ER stress response was detected in MDSCs isolated from cancer patients and tumor-bearing mice, but not in control neutrophils or monocytes, and blockade of ER stress abrogated tumor-associated changes in TRAIL-Rs. Together, these data indicate that MDSC pathophysiology is linked to ER stress, which shortens the lifespan of these cells in the periphery and promotes expansion in BM. Furthermore, TRAIL-Rs can be considered as potential targets for selectively inhibiting MDSCs.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Norris, B. A., et al (2013). "Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity" Immunity 38(2): 309-321.
PubMed
Resolution of acute and chronic viral infections requires activation of innate cells to initiate and maintain adaptive immune responses. Here we report that infection with acute Armstrong (ARM) or chronic Clone 13 (C13) strains of lymphocytic choriomeningitis virus (LCMV) led to two distinct phases of innate immune response. During the first 72 hr of infection, dendritic cells upregulated activation markers and stimulated antiviral CD8(+) T cells, independent of viral strain. Seven days after infection, there was an increase in Ly6C(hi) monocytic and Gr-1(hi) neutrophilic cells in lymphoid organs and blood. This expansion in cell numbers was enhanced and sustained in C13 infection, whereas it occurred only transiently with ARM infection. These cells resembled myeloid-derived suppressor cells and potently suppressed T cell proliferation. The reduction of monocytic cells in Ccr2(-/-) mice or after Gr-1 antibody depletion enhanced antiviral T cell function. Thus, innate cells have an important immunomodulatory role throughout chronic infection.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
van der Merwe, M., et al (2013). "Recipient myeloid-derived immunomodulatory cells induce PD-1 ligand-dependent donor CD4+Foxp3+ regulatory T cell proliferation and donor-recipient immune tolerance after murine nonmyeloablative bone marrow transplant
PubMed
We showed previously that nonmyeloablative total lymphoid irradiation/rabbit anti-thymocyte serum (TLI/ATS) conditioning facilitates potent donor-recipient immune tolerance following bone marrow transplantation (BMT) across MHC barriers via recipient invariant NKT (iNKT) cell-derived IL-4-dependent expansion of donor Foxp3(+) naturally occurring regulatory T cells (nTregs). In this study, we report a more specific mechanism. Wild-type (WT) BALB/c (H-2(d)) hosts were administered TLI/ATS and BMT from WT or STAT6(-/-) C57BL/6 (H-2(b)) donors. Following STAT6(-/-) BMT, donor nTregs demonstrated no loss of proliferation in vivo, indicating that an IL-4-responsive population in the recipient, rather than the donor, drives donor nTreg proliferation. In graft-versus-host disease (GVHD) target organs, three recipient CD11b(+) cell subsets (Gr-1(high)CD11c(-), Gr-1(int)CD11c(-), and Gr-1(low)CD11c(+)) were enriched early after TLI/ATS + BMT versus total body irradiation/ATS + BMT. Gr-1(low)CD11c(+) cells induced potent H-2K(b+)CD4(+)Foxp3(+) nTreg proliferation in vitro in 72-h MLRs. Gr-1(low)CD11c(+) cells were reduced significantly in STAT6(-/-) and iNKT cell-deficient Jalpha18(-/-) BALB/c recipients after TLI/ATS + BMT. Depletion of CD11b(+) cells resulted in severe acute GVHD, and adoptive transfer of WT Gr-1(low)CD11c(+) cells to Jalpha18(-/-) BALB/c recipients of TLI/ATS + BMT restored day-6 donor Foxp3(+) nTreg proliferation and protection from CD8 effector T cell-mediated GVHD. Blockade of programmed death ligand 1 and 2, but not CD40, TGF-beta signaling, arginase 1, or iNOS, inhibited nTreg proliferation in cocultures of recipient-derived Gr-1(low)CD11c(+) cells with donor nTregs. Through iNKT-dependent Th2 polarization, myeloid-derived immunomodulatory dendritic cells are expanded after nonmyeloablative TLI/ATS conditioning and allogeneic BMT, induce PD-1 ligand-dependent donor nTreg proliferation, and maintain potent graft-versus-host immune tolerance.
in vivo depletion of Gr-1+ myeloid cells
Ordonez-Rueda, D., et al (2012). "A hypomorphic mutation in the Gfi1 transcriptional repressor results in a novel form of neutropenia" Eur J Immunol 42(9): 2395-2408.
PubMed
Using N-ethyl-N-nitrosourea-induced mutagenesis, we established a mouse model with a novel form of neutropenia resulting from a point mutation in the transcriptional repressor Growth Factor Independence 1 (Gfi1). These mice, called Genista, had normal viability and no weight loss, in contrast to mice expressing null alleles of the Gfi1 gene. Furthermore, the Genista mutation had a very limited impact on lymphopoiesis or on T- and B-cell function. Within the bone marrow (BM), the Genista mutation resulted in a slight increase of monopoiesis and in a block of terminal granulopoiesis. This block occurred just after the metamyelocytic stage and resulted in the generation of small numbers of atypical CD11b(+) Ly-6G(int) neutrophils, the nuclear morphology of which resembled that of mature WT neutrophils. Unexpectedly, once released from the BM, these atypical neutrophils contributed to induce mild forms of autoantibody-induced arthritis and of immune complex-mediated lung alveolitis. They additionally failed to provide resistance to acute bacterial infection. Our study demonstrates that a hypomorphic mutation in the Gfi1 transcriptional repressor results in a novel form of neutropenia characterized by a split pattern of functional responses, reflecting the distinct thresholds required for eliciting neutrophil-mediated inflammatory and anti-infectious responses.
in vivo depletion of Gr-1+ myeloid cells
in vivo neutrophil depletion
Carr, K. D., et al (2011). "Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection" Eur J Immunol 41(9): 2666-2676.
PubMed
Previous studies have suggested that neutrophils are required for resistance during infection with multiple pathogenic microorganisms. However, the depleting antibody used in those studies binds to both Ly6G and Ly6C (anti-Gr-1; clone RB6-8C5). This antibody has been shown to deplete not only neutrophils but also monocytes and a subset of CD8(+) T cells. Recently, an antibody against Ly6G, which specifically depletes neutrophils, was characterized. In the present study, neutrophils are depleted using the antibody against Ly6G during infection with the intracellular bacterium Listeria monocytogenes (LM). Our data show that neutrophil-depleted mice are much less susceptible to infection than mice depleted with anti-Gr-1. Although neutrophils are required for clearance of LM, their importance is more pronounced in the liver and during a high-dose bacterial challenge. Furthermore, we demonstrate that the protection mediated by neutrophils is due to the production of TNF-alpha, but not IFN-gamma. Additionally, neutrophils are not required for the recruitment of monocytes or the generation of adaptive T-cell responses during LM infection. This study highlights the importance of neutrophils during LM infection, and indicate that depletion of neutrophils is less detrimental to the host than depletion of all Gr-1-expressing cell populations.
in vivo depletion of Gr-1+ myeloid cells
Waight, J. D., et al (2011). "Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism" PLoS One 6(11): e27690.
PubMed
Myeloid-derived suppressor cells (MDSC) are induced under diverse pathologic conditions, including neoplasia, and suppress innate and adaptive immunity. While the mechanisms by which MDSC mediate immunosuppression are well-characterized, details on how they develop remain less understood. This is complicated further by the fact that MDSC comprise multiple myeloid cell types, namely monocytes and granulocytes, reflecting diverse stages of differentiation and the proportion of these subpopulations vary among different neoplastic models. Thus, it is thought that the type and quantities of inflammatory mediators generated during neoplasia dictate the composition of the resultant MDSC response. Although much interest has been devoted to monocytic MDSC biology, a fundamental gap remains in our understanding of the derivation of granulocytic MDSC. In settings of heightened granulocytic MDSC responses, we hypothesized that inappropriate production of G-CSF is a key initiator of granulocytic MDSC accumulation. We observed abundant amounts of G-CSF in vivo, which correlated with robust granulocytic MDSC responses in multiple tumor models. Using G-CSF loss- and gain-of-function approaches, we demonstrated for the first time that: 1) abrogating G-CSF production significantly diminished granulocytic MDSC accumulation and tumor growth; 2) ectopically over-expressing G-CSF in G-CSF-negative tumors significantly augmented granulocytic MDSC accumulation and tumor growth; and 3) treatment of naive healthy mice with recombinant G-CSF protein elicited granulocytic-like MDSC remarkably similar to those induced under tumor-bearing conditions. Collectively, we demonstrated that tumor-derived G-CSF enhances tumor growth through granulocytic MDSC-dependent mechanisms. These findings provide us with novel insights into MDSC subset development and potentially new biomarkers or targets for cancer therapy.
Immunohistochemistry (paraffin)
Li, M., et al (2006). "Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis" Proc Natl Acad Sci U S A 103(31): 11736-11741.
PubMed
We have demonstrated that cytokine thymic stromal lymphopoietin (TSLP), whose expression is rapidly induced upon keratinocyte-selective ablation of retinoid X receptors (RXRs) -alpha and -beta in the mouse (RXRalphabeta(ep-/-) mice), plays a key role in initiating a skin and systemic atopic dermatitis-like phenotype. We show here that topical application of the physiologically active ligand [1alpha,25-(OH)(2)D(3); calcitriol] of the vitamin D receptor, or of its low-calcemic analog MC903 (calcipotriol; Dovonex), induces TSLP expression in epidermal keratinocytes, which results in an atopic dermatitis-like syndrome mimicking that seen in RXRalphabeta(ep-/-) mutants and transgenic mice overexpressing TSLP in keratinocytes. Furthermore, topical application of retinoic acid receptor RARgamma-selective agonist BMS961 also induces TSLP expression either on its own or synergistically with 1alpha,25-(OH)(2)D(3). Our data demonstrate that RXR/vitamin D receptor and RXR/retinoic acid receptor-gamma heterodimers and their ligands cell-autonomously control the expression of TSLP in epidermal keratinocytes of the mouse. We propose molecular mechanisms through which vitamin D3 and retinoic acid signalings could be involved in the pathogenesis of atopic diseases.
Immunohistochemistry (frozen)
Brown, C. R., et al (2004). "Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental lyme arthritis via the recruitment of Gr-1- polymorphonuclear leukocyte-like cells" Infect Immun 72(9): 4956-49
PubMed
Recently, we demonstrated that blocking the entry of neutrophils into Borrelia burgdorferi-infected joints in mice deficient in the chemokine receptor CXCR2 prevented the development of experimental Lyme arthritis. Neutrophils were marginalized in blood vessels at the site of infection but could not enter the joint tissue. In the present study, we treated both genetically arthritis-resistant DBA/2J (DBA) and arthritis-susceptible C3H/HeJ (C3H) mice with the neutrophil-depleting monoclonal antibody RB6-8C5 (RB6) to determine the effect on arthritis development. Surprisingly, both DBA and C3H mice treated with RB6 developed arthritis at 1 week postinfection, approximately 1 week earlier than the control-treated C3H mice. The early development of arthritis in the RB6-treated mice was accompanied by an influx into the joints of cells with ring-shaped polymorphonuclear leukocyte (PMN) cell morphology that were negative for the Gr-1 neutrophil maturation marker. RB6 treatment of mice also resulted in increased numbers of B. burgdorferi cells in the joints at 7 days postinfection and earlier expression of the chemokines KC and monocyte chemoattractant protein 1 in the joints compared to control-treated animals. Together, these results suggest that recruitment of neutrophils or PMN-like cells into an infected joint is a key requirement for Lyme arthritis development and that altered recruitment of these cells into the joints of arthritis-resistant mice can exacerbate the development of pathology.
in vivo depletion of Gr-1+ myeloid cells
in vivo neutrophil depletion
Carr, K. D., et al (2011). "Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection" Eur J Immunol 41(9): 2666-2676.
PubMed
Previous studies have suggested that neutrophils are required for resistance during infection with multiple pathogenic microorganisms. However, the depleting antibody used in those studies binds to both Ly6G and Ly6C (anti-Gr-1; clone RB6-8C5). This antibody has been shown to deplete not only neutrophils but also monocytes and a subset of CD8(+) T cells. Recently, an antibody against Ly6G, which specifically depletes neutrophils, was characterized. In the present study, neutrophils are depleted using the antibody against Ly6G during infection with the intracellular bacterium Listeria monocytogenes (LM). Our data show that neutrophil-depleted mice are much less susceptible to infection than mice depleted with anti-Gr-1. Although neutrophils are required for clearance of LM, their importance is more pronounced in the liver and during a high-dose bacterial challenge. Furthermore, we demonstrate that the protection mediated by neutrophils is due to the production of TNF-alpha, but not IFN-gamma. Additionally, neutrophils are not required for the recruitment of monocytes or the generation of adaptive T-cell responses during LM infection. This study highlights the importance of neutrophils during LM infection, and indicate that depletion of neutrophils is less detrimental to the host than depletion of all Gr-1-expressing cell populations.
in vivo IL-17A neutralization
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
in vivo TNFα neutralization
Flow Cytometry
in vivo IL-6 neutralization
in vivo IL-1β neutralization
Khmaladze, I., et al (2014). "Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice" Proc Natl Acad Sci U S A 111(35): E3669-3678.
PubMed
Psoriasis (Ps) and psoriasis arthritis (PsA) are poorly understood common diseases, induced by unknown environmental factors, affecting skin and articular joints. A single i.p. exposure to mannan from Saccharomyces cerevisiae induced an acute inflammation in inbred mouse strains resembling human Ps and PsA-like disease, whereas multiple injections induced a relapsing disease. Exacerbation of disease severity was observed in mice deficient for generation of reactive oxygen species (ROS). Interestingly, restoration of ROS production, specifically in macrophages, ameliorated both skin and joint disease. Neutralization of IL-17A, mainly produced by gammadelta T cells, completely blocked disease symptoms. Furthermore, mice depleted of granulocytes were resistant to disease development. In contrast, certain acute inflammatory mediators (C5, Fcgamma receptor III, mast cells, and histamine) and adaptive immune players (alphabeta T and B cells) were redundant in disease induction. Hence, we propose that mannan-induced activation of macrophages leads to TNF-alpha secretion and stimulation of local gammadelta T cells secreting IL-17A. The combined action of activated macrophages and IL-17A produced in situ drives neutrophil infiltration in the epidermis and dermis of the skin, leading to disease manifestations. Thus, our finding suggests a new mechanism triggered by exposure to exogenous microbial components, such as mannan, that can induce and exacerbate Ps and PsA.
in vivo depletion of Gr-1+ myeloid cells
Bansal, S., et al (2018). "IL-1 Signaling Prevents Alveolar Macrophage Depletion during Influenza and Streptococcus pneumoniae Coinfection" J Immunol 200(4): 1425-1433.
PubMed
Influenza and bacterial coinfection is a significant cause of hospitalization and death in humans during influenza epidemics and pandemics. However, the fundamental protective and pathogenic mechanisms involved in this complex virus-host-bacterium interaction remain incompletely understood. In this study, we have developed mild to lethal influenza and Streptococcus pneumoniae coinfection models for comparative analyses of disease pathogenesis. Specifically, wild-type and IL-1R type 1-deficient (Il1r1(-/-) ) mice were infected with influenza virus and then superchallenged with noninvasive S. pneumoniae serotype 14 (Spn14) or S. pneumoniae serotype 19A (Spn19A). The coinfections were followed by comparative analyses of inflammatory responses and animal protection. We found that resident alveolar macrophages are efficient in the clearance of both pneumococcal serotypes in the absence of influenza infection; in contrast, they are essential for airway control of Spn14 infection but not Spn19A infection. In agreement, TNF-alpha and neutrophils play a compensatory protective role in secondary bacterial infection associated with Spn19A; however, the essential requirement for alveolar macrophage-mediated clearance significantly enhances the virulence of Spn14 during postinfluenza pneumococcal infection. Furthermore, we show that, although IL-1 signaling is not required for host defense against pneumococcal infection alone, it is essential for sustaining antibacterial immunity during postinfluenza pneumococcal infection, as evidenced by significantly aggravated bacterial burden and animal mortality in Il1r1(-/-) mice. Mechanistically, we show that through preventing alveolar macrophage depletion, inflammatory cytokine IL-1 signaling is critically involved in host resistance to influenza and pneumococcal coinfection.
in vivo depletion of Gr-1+ myeloid cells
Dahlgren, M. W., et al (2015). "T follicular helper, but not Th1, cell differentiation in the absence of conventional dendritic cells" J Immunol 194(11): 5187-5199.
PubMed
Development of long-lived humoral immunity is dependent on CXCR5-expressing T follicular helper (Tfh) cells, which develop concomitantly to effector Th cells that support cellular immunity. Conventional dendritic cells (cDCs) are critical APCs for initial priming of naive CD4(+) T cells but, importantly, also provide accessory signals that govern effector Th cell commitment. To define the accessory role of cDCs during the concurrent development of Tfh and effector Th1 cells, we performed high-dose Ag immunization in conjunction with the Th1-biased adjuvant polyinosinic:polycytidylic acid (pI:C). In the absence of cDCs, pI:C failed to induce Th1 cell commitment and IgG2c production. However, cDC depletion did not impair Tfh cell differentiation or germinal center formation, and long-lived IgG1 responses of unaltered affinity developed in mice lacking cDCs at the time point for immunization. Thus, cDCs are required for the pI:C-driven Th1 cell fate commitment but have no crucial accessory function in relation to Tfh cell differentiation.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Wang, H., et al (2015). "P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy" Cell Res 25(6): 674-690.
PubMed
Septic encephalopathy (SE) is a critical factor determining sepsis mortality. Vascular inflammation is known to be involved in SE, but the molecular events that lead to the development of encephalopathy remain unclear. Using time-lapse in vivo two-photon laser scanning microscopy, we provide the first direct evidence that cecal ligation and puncture in septic mice induces microglial trafficking to sites adjacent to leukocyte adhesion on inflamed cerebral microvessels. Our data further demonstrate that septic injury increased the chemokine CXCL1 level in brain endothelial cells by activating endothelial P2RX7 and eventually enhanced the binding of Mac-1 (CD11b/CD18)-expressing leukocytes to endothelial ICAM-1. In turn, leukocyte adhesion upregulated endothelial CX3CL1, thereby triggering microglia trafficking to the injured site. The sepsis-induced increase in endothelial CX3CL1 was abolished in CD18 hypomorphic mutant mice. Inhibition of the P2RX7 pathway not only decreased endothelial ICAM-1 expression and leukocyte adhesion but also prevented microglia overactivation, reduced brain injury, and consequently doubled the early survival of septic mice. These results demonstrate the role of the P2RX7 pathway in linking neurovascular inflammation to brain damage in vivo and provide a rationale for targeting endothelial P2RX7 for neurovascular protection during SE.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Bryant, J., et al (2014). "Preemptive donor apoptotic cell infusions induce IFN-gamma-producing myeloid-derived suppressor cells for cardiac allograft protection" J Immunol 192(12): 6092-6101.
PubMed
We have previously shown that preemptive infusion of apoptotic donor splenocytes treated with the chemical cross-linker ethylcarbodiimide (ECDI-SPs) induces long-term allograft survival in full MHC-mismatched models of allogeneic islet and cardiac transplantation. The role of myeloid-derived suppressor cells (MDSCs) in the graft protection provided by ECDI-SPs is unclear. In this study, we demonstrate that infusions of ECDI-SPs increase two populations of CD11b(+) cells in the spleen that phenotypically resemble monocytic-like (CD11b(+)Ly6C(high)) and granulocytic-like (CD11b(+)Gr1(high)) MDSCs. Both populations suppress T cell proliferation in vitro and traffic to the cardiac allografts in vivo to mediate their protection via inhibition of local CD8 T cell accumulation and potentially also via induction and homing of regulatory T cells. Importantly, repeated treatments with ECDI-SPs induce the CD11b(+)Gr1(high) cells to produce a high level of IFN-gamma and to exhibit an enhanced responsiveness to IFN-gamma by expressing higher levels of downstream effector molecules ido and nos2. Consequently, neutralization of IFN-gamma completely abolishes the suppressive capacity of this population. We conclude that donor ECDI-SPs induce the expansion of two populations of MDSCs important for allograft protection mediated in part by intrinsic IFN-gamma-dependent mechanisms. This form of preemptive donor apoptotic cell infusions has significant potential for the therapeutic manipulation of MDSCs for transplant tolerance induction.
in vivo depletion of Gr-1+ myeloid cells
in vivo CTLA-4 neutralization
in vivo induction TRAIL-mediated apoptosis
in vitro induction TRAIL-mediated apoptosis
Condamine, T., et al (2014). "ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis" J Clin Invest 124(6): 2626-2639.
PubMed
Myeloid-derived suppressor cells (MDSCs) dampen the immune response thorough inhibition of T cell activation and proliferation and often are expanded in pathological conditions. Here, we studied the fate of MDSCs in cancer. Unexpectedly, MDSCs had lower viability and a shorter half-life in tumor-bearing mice compared with neutrophils and monocytes. The reduction of MDSC viability was due to increased apoptosis, which was mediated by increased expression of TNF-related apoptosis-induced ligand receptors (TRAIL-Rs) in these cells. Targeting TRAIL-Rs in naive mice did not affect myeloid cell populations, but it dramatically reduced the presence of MDSCs and improved immune responses in tumor-bearing mice. Treatment of myeloid cells with proinflammatory cytokines did not affect TRAIL-R expression; however, induction of ER stress in myeloid cells recapitulated changes in TRAIL-R expression observed in tumor-bearing hosts. The ER stress response was detected in MDSCs isolated from cancer patients and tumor-bearing mice, but not in control neutrophils or monocytes, and blockade of ER stress abrogated tumor-associated changes in TRAIL-Rs. Together, these data indicate that MDSC pathophysiology is linked to ER stress, which shortens the lifespan of these cells in the periphery and promotes expansion in BM. Furthermore, TRAIL-Rs can be considered as potential targets for selectively inhibiting MDSCs.
in vivo IL-17A neutralization
in vivo depletion of Gr-1+ myeloid cells
in vivo ILC depletion
Ermann, J., et al (2014). "Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice" Proc Natl Acad Sci U S A 111(25): E2559-2566.
PubMed
T-bet(-/-).Rag2(-/-) (TRUC) mice spontaneously develop microbiota-driven, TNF-mediated large bowel inflammation that resembles human ulcerative colitis. We show here that IL-23 and IL-1-dependent secretion of IL-17A by innate lymphoid cells (ILCs; defined as CD45(+)lin(-)Thy1(hi)NKp46(-)) is a second critical pathway in this model. Using an in vitro coculture system of bone marrow-derived dendritic cells (DCs) and freshly isolated FACS-purified ILCs, we demonstrate that IL-23 and IL-1 secreted by DCs in response to microbial stimulation work together to induce IL-17A production by ILCs. TNF is not required for IL-17A secretion by ILCs in vitro but synergizes with IL-17A to induce the expression of neutrophil-attracting chemokines. Upstream, activation of the IL-23/IL-17A axis is regulated by nucleotide-binding oligomerization domain containing (Nod)/receptor-interacting serine-threonine kinase 2 (Ripk2) signals in DCs. Genetic ablation of the Nod/Ripk2 signaling pathway protects TRUC mice from developing colitis without affecting the colitogenicity of the intestinal microbiota. Our data provide insight into the complex network of interactions between IL-17A-secreting ILCs and other components of the innate immune system in the development of colitis.
in vivo IL-17A neutralization
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
in vivo TNFα neutralization
Flow Cytometry
in vivo IL-6 neutralization
in vivo IL-1β neutralization
Khmaladze, I., et al (2014). "Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice" Proc Natl Acad Sci U S A 111(35): E3669-3678.
PubMed
Psoriasis (Ps) and psoriasis arthritis (PsA) are poorly understood common diseases, induced by unknown environmental factors, affecting skin and articular joints. A single i.p. exposure to mannan from Saccharomyces cerevisiae induced an acute inflammation in inbred mouse strains resembling human Ps and PsA-like disease, whereas multiple injections induced a relapsing disease. Exacerbation of disease severity was observed in mice deficient for generation of reactive oxygen species (ROS). Interestingly, restoration of ROS production, specifically in macrophages, ameliorated both skin and joint disease. Neutralization of IL-17A, mainly produced by gammadelta T cells, completely blocked disease symptoms. Furthermore, mice depleted of granulocytes were resistant to disease development. In contrast, certain acute inflammatory mediators (C5, Fcgamma receptor III, mast cells, and histamine) and adaptive immune players (alphabeta T and B cells) were redundant in disease induction. Hence, we propose that mannan-induced activation of macrophages leads to TNF-alpha secretion and stimulation of local gammadelta T cells secreting IL-17A. The combined action of activated macrophages and IL-17A produced in situ drives neutrophil infiltration in the epidermis and dermis of the skin, leading to disease manifestations. Thus, our finding suggests a new mechanism triggered by exposure to exogenous microbial components, such as mannan, that can induce and exacerbate Ps and PsA.
in vivo depletion of Gr-1+ myeloid cells
Bansal, S., et al (2018). "IL-1 Signaling Prevents Alveolar Macrophage Depletion during Influenza and Streptococcus pneumoniae Coinfection" J Immunol 200(4): 1425-1433.
PubMed
Influenza and bacterial coinfection is a significant cause of hospitalization and death in humans during influenza epidemics and pandemics. However, the fundamental protective and pathogenic mechanisms involved in this complex virus-host-bacterium interaction remain incompletely understood. In this study, we have developed mild to lethal influenza and Streptococcus pneumoniae coinfection models for comparative analyses of disease pathogenesis. Specifically, wild-type and IL-1R type 1-deficient (Il1r1(-/-) ) mice were infected with influenza virus and then superchallenged with noninvasive S. pneumoniae serotype 14 (Spn14) or S. pneumoniae serotype 19A (Spn19A). The coinfections were followed by comparative analyses of inflammatory responses and animal protection. We found that resident alveolar macrophages are efficient in the clearance of both pneumococcal serotypes in the absence of influenza infection; in contrast, they are essential for airway control of Spn14 infection but not Spn19A infection. In agreement, TNF-alpha and neutrophils play a compensatory protective role in secondary bacterial infection associated with Spn19A; however, the essential requirement for alveolar macrophage-mediated clearance significantly enhances the virulence of Spn14 during postinfluenza pneumococcal infection. Furthermore, we show that, although IL-1 signaling is not required for host defense against pneumococcal infection alone, it is essential for sustaining antibacterial immunity during postinfluenza pneumococcal infection, as evidenced by significantly aggravated bacterial burden and animal mortality in Il1r1(-/-) mice. Mechanistically, we show that through preventing alveolar macrophage depletion, inflammatory cytokine IL-1 signaling is critically involved in host resistance to influenza and pneumococcal coinfection.
in vivo depletion of Gr-1+ myeloid cells
Dahlgren, M. W., et al (2015). "T follicular helper, but not Th1, cell differentiation in the absence of conventional dendritic cells" J Immunol 194(11): 5187-5199.
PubMed
Development of long-lived humoral immunity is dependent on CXCR5-expressing T follicular helper (Tfh) cells, which develop concomitantly to effector Th cells that support cellular immunity. Conventional dendritic cells (cDCs) are critical APCs for initial priming of naive CD4(+) T cells but, importantly, also provide accessory signals that govern effector Th cell commitment. To define the accessory role of cDCs during the concurrent development of Tfh and effector Th1 cells, we performed high-dose Ag immunization in conjunction with the Th1-biased adjuvant polyinosinic:polycytidylic acid (pI:C). In the absence of cDCs, pI:C failed to induce Th1 cell commitment and IgG2c production. However, cDC depletion did not impair Tfh cell differentiation or germinal center formation, and long-lived IgG1 responses of unaltered affinity developed in mice lacking cDCs at the time point for immunization. Thus, cDCs are required for the pI:C-driven Th1 cell fate commitment but have no crucial accessory function in relation to Tfh cell differentiation.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Wang, H., et al (2015). "P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy" Cell Res 25(6): 674-690.
PubMed
Septic encephalopathy (SE) is a critical factor determining sepsis mortality. Vascular inflammation is known to be involved in SE, but the molecular events that lead to the development of encephalopathy remain unclear. Using time-lapse in vivo two-photon laser scanning microscopy, we provide the first direct evidence that cecal ligation and puncture in septic mice induces microglial trafficking to sites adjacent to leukocyte adhesion on inflamed cerebral microvessels. Our data further demonstrate that septic injury increased the chemokine CXCL1 level in brain endothelial cells by activating endothelial P2RX7 and eventually enhanced the binding of Mac-1 (CD11b/CD18)-expressing leukocytes to endothelial ICAM-1. In turn, leukocyte adhesion upregulated endothelial CX3CL1, thereby triggering microglia trafficking to the injured site. The sepsis-induced increase in endothelial CX3CL1 was abolished in CD18 hypomorphic mutant mice. Inhibition of the P2RX7 pathway not only decreased endothelial ICAM-1 expression and leukocyte adhesion but also prevented microglia overactivation, reduced brain injury, and consequently doubled the early survival of septic mice. These results demonstrate the role of the P2RX7 pathway in linking neurovascular inflammation to brain damage in vivo and provide a rationale for targeting endothelial P2RX7 for neurovascular protection during SE.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Bryant, J., et al (2014). "Preemptive donor apoptotic cell infusions induce IFN-gamma-producing myeloid-derived suppressor cells for cardiac allograft protection" J Immunol 192(12): 6092-6101.
PubMed
We have previously shown that preemptive infusion of apoptotic donor splenocytes treated with the chemical cross-linker ethylcarbodiimide (ECDI-SPs) induces long-term allograft survival in full MHC-mismatched models of allogeneic islet and cardiac transplantation. The role of myeloid-derived suppressor cells (MDSCs) in the graft protection provided by ECDI-SPs is unclear. In this study, we demonstrate that infusions of ECDI-SPs increase two populations of CD11b(+) cells in the spleen that phenotypically resemble monocytic-like (CD11b(+)Ly6C(high)) and granulocytic-like (CD11b(+)Gr1(high)) MDSCs. Both populations suppress T cell proliferation in vitro and traffic to the cardiac allografts in vivo to mediate their protection via inhibition of local CD8 T cell accumulation and potentially also via induction and homing of regulatory T cells. Importantly, repeated treatments with ECDI-SPs induce the CD11b(+)Gr1(high) cells to produce a high level of IFN-gamma and to exhibit an enhanced responsiveness to IFN-gamma by expressing higher levels of downstream effector molecules ido and nos2. Consequently, neutralization of IFN-gamma completely abolishes the suppressive capacity of this population. We conclude that donor ECDI-SPs induce the expansion of two populations of MDSCs important for allograft protection mediated in part by intrinsic IFN-gamma-dependent mechanisms. This form of preemptive donor apoptotic cell infusions has significant potential for the therapeutic manipulation of MDSCs for transplant tolerance induction.
in vivo depletion of Gr-1+ myeloid cells
in vivo CTLA-4 neutralization
in vivo induction TRAIL-mediated apoptosis
in vitro induction TRAIL-mediated apoptosis
Condamine, T., et al (2014). "ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis" J Clin Invest 124(6): 2626-2639.
PubMed
Myeloid-derived suppressor cells (MDSCs) dampen the immune response thorough inhibition of T cell activation and proliferation and often are expanded in pathological conditions. Here, we studied the fate of MDSCs in cancer. Unexpectedly, MDSCs had lower viability and a shorter half-life in tumor-bearing mice compared with neutrophils and monocytes. The reduction of MDSC viability was due to increased apoptosis, which was mediated by increased expression of TNF-related apoptosis-induced ligand receptors (TRAIL-Rs) in these cells. Targeting TRAIL-Rs in naive mice did not affect myeloid cell populations, but it dramatically reduced the presence of MDSCs and improved immune responses in tumor-bearing mice. Treatment of myeloid cells with proinflammatory cytokines did not affect TRAIL-R expression; however, induction of ER stress in myeloid cells recapitulated changes in TRAIL-R expression observed in tumor-bearing hosts. The ER stress response was detected in MDSCs isolated from cancer patients and tumor-bearing mice, but not in control neutrophils or monocytes, and blockade of ER stress abrogated tumor-associated changes in TRAIL-Rs. Together, these data indicate that MDSC pathophysiology is linked to ER stress, which shortens the lifespan of these cells in the periphery and promotes expansion in BM. Furthermore, TRAIL-Rs can be considered as potential targets for selectively inhibiting MDSCs.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Norris, B. A., et al (2013). "Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity" Immunity 38(2): 309-321.
PubMed
Resolution of acute and chronic viral infections requires activation of innate cells to initiate and maintain adaptive immune responses. Here we report that infection with acute Armstrong (ARM) or chronic Clone 13 (C13) strains of lymphocytic choriomeningitis virus (LCMV) led to two distinct phases of innate immune response. During the first 72 hr of infection, dendritic cells upregulated activation markers and stimulated antiviral CD8(+) T cells, independent of viral strain. Seven days after infection, there was an increase in Ly6C(hi) monocytic and Gr-1(hi) neutrophilic cells in lymphoid organs and blood. This expansion in cell numbers was enhanced and sustained in C13 infection, whereas it occurred only transiently with ARM infection. These cells resembled myeloid-derived suppressor cells and potently suppressed T cell proliferation. The reduction of monocytic cells in Ccr2(-/-) mice or after Gr-1 antibody depletion enhanced antiviral T cell function. Thus, innate cells have an important immunomodulatory role throughout chronic infection.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
van der Merwe, M., et al (2013). "Recipient myeloid-derived immunomodulatory cells induce PD-1 ligand-dependent donor CD4+Foxp3+ regulatory T cell proliferation and donor-recipient immune tolerance after murine nonmyeloablative bone marrow transplant
PubMed
We showed previously that nonmyeloablative total lymphoid irradiation/rabbit anti-thymocyte serum (TLI/ATS) conditioning facilitates potent donor-recipient immune tolerance following bone marrow transplantation (BMT) across MHC barriers via recipient invariant NKT (iNKT) cell-derived IL-4-dependent expansion of donor Foxp3(+) naturally occurring regulatory T cells (nTregs). In this study, we report a more specific mechanism. Wild-type (WT) BALB/c (H-2(d)) hosts were administered TLI/ATS and BMT from WT or STAT6(-/-) C57BL/6 (H-2(b)) donors. Following STAT6(-/-) BMT, donor nTregs demonstrated no loss of proliferation in vivo, indicating that an IL-4-responsive population in the recipient, rather than the donor, drives donor nTreg proliferation. In graft-versus-host disease (GVHD) target organs, three recipient CD11b(+) cell subsets (Gr-1(high)CD11c(-), Gr-1(int)CD11c(-), and Gr-1(low)CD11c(+)) were enriched early after TLI/ATS + BMT versus total body irradiation/ATS + BMT. Gr-1(low)CD11c(+) cells induced potent H-2K(b+)CD4(+)Foxp3(+) nTreg proliferation in vitro in 72-h MLRs. Gr-1(low)CD11c(+) cells were reduced significantly in STAT6(-/-) and iNKT cell-deficient Jalpha18(-/-) BALB/c recipients after TLI/ATS + BMT. Depletion of CD11b(+) cells resulted in severe acute GVHD, and adoptive transfer of WT Gr-1(low)CD11c(+) cells to Jalpha18(-/-) BALB/c recipients of TLI/ATS + BMT restored day-6 donor Foxp3(+) nTreg proliferation and protection from CD8 effector T cell-mediated GVHD. Blockade of programmed death ligand 1 and 2, but not CD40, TGF-beta signaling, arginase 1, or iNOS, inhibited nTreg proliferation in cocultures of recipient-derived Gr-1(low)CD11c(+) cells with donor nTregs. Through iNKT-dependent Th2 polarization, myeloid-derived immunomodulatory dendritic cells are expanded after nonmyeloablative TLI/ATS conditioning and allogeneic BMT, induce PD-1 ligand-dependent donor nTreg proliferation, and maintain potent graft-versus-host immune tolerance.
in vivo depletion of Gr-1+ myeloid cells
Ordonez-Rueda, D., et al (2012). "A hypomorphic mutation in the Gfi1 transcriptional repressor results in a novel form of neutropenia" Eur J Immunol 42(9): 2395-2408.
PubMed
Using N-ethyl-N-nitrosourea-induced mutagenesis, we established a mouse model with a novel form of neutropenia resulting from a point mutation in the transcriptional repressor Growth Factor Independence 1 (Gfi1). These mice, called Genista, had normal viability and no weight loss, in contrast to mice expressing null alleles of the Gfi1 gene. Furthermore, the Genista mutation had a very limited impact on lymphopoiesis or on T- and B-cell function. Within the bone marrow (BM), the Genista mutation resulted in a slight increase of monopoiesis and in a block of terminal granulopoiesis. This block occurred just after the metamyelocytic stage and resulted in the generation of small numbers of atypical CD11b(+) Ly-6G(int) neutrophils, the nuclear morphology of which resembled that of mature WT neutrophils. Unexpectedly, once released from the BM, these atypical neutrophils contributed to induce mild forms of autoantibody-induced arthritis and of immune complex-mediated lung alveolitis. They additionally failed to provide resistance to acute bacterial infection. Our study demonstrates that a hypomorphic mutation in the Gfi1 transcriptional repressor results in a novel form of neutropenia characterized by a split pattern of functional responses, reflecting the distinct thresholds required for eliciting neutrophil-mediated inflammatory and anti-infectious responses.
in vivo depletion of Gr-1+ myeloid cells
Waight, J. D., et al (2011). "Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism" PLoS One 6(11): e27690.
PubMed
Myeloid-derived suppressor cells (MDSC) are induced under diverse pathologic conditions, including neoplasia, and suppress innate and adaptive immunity. While the mechanisms by which MDSC mediate immunosuppression are well-characterized, details on how they develop remain less understood. This is complicated further by the fact that MDSC comprise multiple myeloid cell types, namely monocytes and granulocytes, reflecting diverse stages of differentiation and the proportion of these subpopulations vary among different neoplastic models. Thus, it is thought that the type and quantities of inflammatory mediators generated during neoplasia dictate the composition of the resultant MDSC response. Although much interest has been devoted to monocytic MDSC biology, a fundamental gap remains in our understanding of the derivation of granulocytic MDSC. In settings of heightened granulocytic MDSC responses, we hypothesized that inappropriate production of G-CSF is a key initiator of granulocytic MDSC accumulation. We observed abundant amounts of G-CSF in vivo, which correlated with robust granulocytic MDSC responses in multiple tumor models. Using G-CSF loss- and gain-of-function approaches, we demonstrated for the first time that: 1) abrogating G-CSF production significantly diminished granulocytic MDSC accumulation and tumor growth; 2) ectopically over-expressing G-CSF in G-CSF-negative tumors significantly augmented granulocytic MDSC accumulation and tumor growth; and 3) treatment of naive healthy mice with recombinant G-CSF protein elicited granulocytic-like MDSC remarkably similar to those induced under tumor-bearing conditions. Collectively, we demonstrated that tumor-derived G-CSF enhances tumor growth through granulocytic MDSC-dependent mechanisms. These findings provide us with novel insights into MDSC subset development and potentially new biomarkers or targets for cancer therapy.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
in vitro TGFβ neutralization
Bodogai, M., et al (2015). "Immunosuppressive and Prometastatic Functions of Myeloid-Derived Suppressive Cells Rely upon Education from Tumor-Associated B Cells" Cancer Res 75(17): 3456-3465.
PubMed
Myeloid-derived suppressive cells (MDSC) have been reported to promote metastasis, but the loss of cancer-induced B cells/B regulatory cells (tBreg) can block metastasis despite MDSC expansion in cancer. Here, using multiple murine tumor models and human MDSC, we show that MDSC populations that expand in cancer have only partially primed regulatory function and limited prometastatic activity unless they are fully educated by tBregs. Cancer-induced tBregs directly activate the regulatory function of both the monocyte and granulocyte subpopulations of MDSC, relying, in part, on TgfbetaR1/TgfbetaR2 signaling. MDSC fully educated in this manner exhibit an increased production of reactive oxygen species and NO and more efficiently suppress CD4(+) and CD8(+) T cells, thereby promoting tumor growth and metastasis. Thus, loss of tBregs or TgfbetaR deficiency in MDSC is sufficient to disable their suppressive function and to block metastasis. Overall, our data indicate that cancer-induced B cells/B regulatory cells are important regulators of the immunosuppressive and prometastatic functions of MDSC.
in vivo depletion of Gr-1+ myeloid cells
Bansal, S., et al (2018). "IL-1 Signaling Prevents Alveolar Macrophage Depletion during Influenza and Streptococcus pneumoniae Coinfection" J Immunol 200(4): 1425-1433.
PubMed
Influenza and bacterial coinfection is a significant cause of hospitalization and death in humans during influenza epidemics and pandemics. However, the fundamental protective and pathogenic mechanisms involved in this complex virus-host-bacterium interaction remain incompletely understood. In this study, we have developed mild to lethal influenza and Streptococcus pneumoniae coinfection models for comparative analyses of disease pathogenesis. Specifically, wild-type and IL-1R type 1-deficient (Il1r1(-/-) ) mice were infected with influenza virus and then superchallenged with noninvasive S. pneumoniae serotype 14 (Spn14) or S. pneumoniae serotype 19A (Spn19A). The coinfections were followed by comparative analyses of inflammatory responses and animal protection. We found that resident alveolar macrophages are efficient in the clearance of both pneumococcal serotypes in the absence of influenza infection; in contrast, they are essential for airway control of Spn14 infection but not Spn19A infection. In agreement, TNF-alpha and neutrophils play a compensatory protective role in secondary bacterial infection associated with Spn19A; however, the essential requirement for alveolar macrophage-mediated clearance significantly enhances the virulence of Spn14 during postinfluenza pneumococcal infection. Furthermore, we show that, although IL-1 signaling is not required for host defense against pneumococcal infection alone, it is essential for sustaining antibacterial immunity during postinfluenza pneumococcal infection, as evidenced by significantly aggravated bacterial burden and animal mortality in Il1r1(-/-) mice. Mechanistically, we show that through preventing alveolar macrophage depletion, inflammatory cytokine IL-1 signaling is critically involved in host resistance to influenza and pneumococcal coinfection.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Wang, H., et al (2015). "P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy" Cell Res 25(6): 674-690.
PubMed
Septic encephalopathy (SE) is a critical factor determining sepsis mortality. Vascular inflammation is known to be involved in SE, but the molecular events that lead to the development of encephalopathy remain unclear. Using time-lapse in vivo two-photon laser scanning microscopy, we provide the first direct evidence that cecal ligation and puncture in septic mice induces microglial trafficking to sites adjacent to leukocyte adhesion on inflamed cerebral microvessels. Our data further demonstrate that septic injury increased the chemokine CXCL1 level in brain endothelial cells by activating endothelial P2RX7 and eventually enhanced the binding of Mac-1 (CD11b/CD18)-expressing leukocytes to endothelial ICAM-1. In turn, leukocyte adhesion upregulated endothelial CX3CL1, thereby triggering microglia trafficking to the injured site. The sepsis-induced increase in endothelial CX3CL1 was abolished in CD18 hypomorphic mutant mice. Inhibition of the P2RX7 pathway not only decreased endothelial ICAM-1 expression and leukocyte adhesion but also prevented microglia overactivation, reduced brain injury, and consequently doubled the early survival of septic mice. These results demonstrate the role of the P2RX7 pathway in linking neurovascular inflammation to brain damage in vivo and provide a rationale for targeting endothelial P2RX7 for neurovascular protection during SE.
in vivo depletion of Gr-1+ myeloid cells
Dahlgren, M. W., et al (2015). "T follicular helper, but not Th1, cell differentiation in the absence of conventional dendritic cells" J Immunol 194(11): 5187-5199.
PubMed
Development of long-lived humoral immunity is dependent on CXCR5-expressing T follicular helper (Tfh) cells, which develop concomitantly to effector Th cells that support cellular immunity. Conventional dendritic cells (cDCs) are critical APCs for initial priming of naive CD4(+) T cells but, importantly, also provide accessory signals that govern effector Th cell commitment. To define the accessory role of cDCs during the concurrent development of Tfh and effector Th1 cells, we performed high-dose Ag immunization in conjunction with the Th1-biased adjuvant polyinosinic:polycytidylic acid (pI:C). In the absence of cDCs, pI:C failed to induce Th1 cell commitment and IgG2c production. However, cDC depletion did not impair Tfh cell differentiation or germinal center formation, and long-lived IgG1 responses of unaltered affinity developed in mice lacking cDCs at the time point for immunization. Thus, cDCs are required for the pI:C-driven Th1 cell fate commitment but have no crucial accessory function in relation to Tfh cell differentiation.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Bodogai, M., et al (2015). "Immunosuppressive and Prometastatic Functions of Myeloid-Derived Suppressive Cells Rely upon Education from Tumor-Associated B Cells" Cancer Res 75(17): 3456-3465.
PubMed
Myeloid-derived suppressive cells (MDSC) have been reported to promote metastasis, but the loss of cancer-induced B cells/B regulatory cells (tBreg) can block metastasis despite MDSC expansion in cancer. Here, using multiple murine tumor models and human MDSC, we show that MDSC populations that expand in cancer have only partially primed regulatory function and limited prometastatic activity unless they are fully educated by tBregs. Cancer-induced tBregs directly activate the regulatory function of both the monocyte and granulocyte subpopulations of MDSC, relying, in part, on TgfbetaR1/TgfbetaR2 signaling. MDSC fully educated in this manner exhibit an increased production of reactive oxygen species and NO and more efficiently suppress CD4(+) and CD8(+) T cells, thereby promoting tumor growth and metastasis. Thus, loss of tBregs or TgfbetaR deficiency in MDSC is sufficient to disable their suppressive function and to block metastasis. Overall, our data indicate that cancer-induced B cells/B regulatory cells are important regulators of the immunosuppressive and prometastatic functions of MDSC.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Bryant, J., et al (2014). "Preemptive donor apoptotic cell infusions induce IFN-gamma-producing myeloid-derived suppressor cells for cardiac allograft protection" J Immunol 192(12): 6092-6101.
PubMed
We have previously shown that preemptive infusion of apoptotic donor splenocytes treated with the chemical cross-linker ethylcarbodiimide (ECDI-SPs) induces long-term allograft survival in full MHC-mismatched models of allogeneic islet and cardiac transplantation. The role of myeloid-derived suppressor cells (MDSCs) in the graft protection provided by ECDI-SPs is unclear. In this study, we demonstrate that infusions of ECDI-SPs increase two populations of CD11b(+) cells in the spleen that phenotypically resemble monocytic-like (CD11b(+)Ly6C(high)) and granulocytic-like (CD11b(+)Gr1(high)) MDSCs. Both populations suppress T cell proliferation in vitro and traffic to the cardiac allografts in vivo to mediate their protection via inhibition of local CD8 T cell accumulation and potentially also via induction and homing of regulatory T cells. Importantly, repeated treatments with ECDI-SPs induce the CD11b(+)Gr1(high) cells to produce a high level of IFN-gamma and to exhibit an enhanced responsiveness to IFN-gamma by expressing higher levels of downstream effector molecules ido and nos2. Consequently, neutralization of IFN-gamma completely abolishes the suppressive capacity of this population. We conclude that donor ECDI-SPs induce the expansion of two populations of MDSCs important for allograft protection mediated in part by intrinsic IFN-gamma-dependent mechanisms. This form of preemptive donor apoptotic cell infusions has significant potential for the therapeutic manipulation of MDSCs for transplant tolerance induction.
in vivo depletion of Gr-1+ myeloid cells
Condamine, T., et al (2014). "ER stress regulates myeloid-derived suppressor cell fate through TRAIL-R-mediated apoptosis" J Clin Invest 124(6): 2626-2639.
PubMed
Myeloid-derived suppressor cells (MDSCs) dampen the immune response thorough inhibition of T cell activation and proliferation and often are expanded in pathological conditions. Here, we studied the fate of MDSCs in cancer. Unexpectedly, MDSCs had lower viability and a shorter half-life in tumor-bearing mice compared with neutrophils and monocytes. The reduction of MDSC viability was due to increased apoptosis, which was mediated by increased expression of TNF-related apoptosis-induced ligand receptors (TRAIL-Rs) in these cells. Targeting TRAIL-Rs in naive mice did not affect myeloid cell populations, but it dramatically reduced the presence of MDSCs and improved immune responses in tumor-bearing mice. Treatment of myeloid cells with proinflammatory cytokines did not affect TRAIL-R expression; however, induction of ER stress in myeloid cells recapitulated changes in TRAIL-R expression observed in tumor-bearing hosts. The ER stress response was detected in MDSCs isolated from cancer patients and tumor-bearing mice, but not in control neutrophils or monocytes, and blockade of ER stress abrogated tumor-associated changes in TRAIL-Rs. Together, these data indicate that MDSC pathophysiology is linked to ER stress, which shortens the lifespan of these cells in the periphery and promotes expansion in BM. Furthermore, TRAIL-Rs can be considered as potential targets for selectively inhibiting MDSCs.
in vivo depletion of Gr-1+ myeloid cells
Ermann, J., et al (2014). "Nod/Ripk2 signaling in dendritic cells activates IL-17A-secreting innate lymphoid cells and drives colitis in T-bet-/-.Rag2-/- (TRUC) mice" Proc Natl Acad Sci U S A 111(25): E2559-2566.
PubMed
T-bet(-/-).Rag2(-/-) (TRUC) mice spontaneously develop microbiota-driven, TNF-mediated large bowel inflammation that resembles human ulcerative colitis. We show here that IL-23 and IL-1-dependent secretion of IL-17A by innate lymphoid cells (ILCs; defined as CD45(+)lin(-)Thy1(hi)NKp46(-)) is a second critical pathway in this model. Using an in vitro coculture system of bone marrow-derived dendritic cells (DCs) and freshly isolated FACS-purified ILCs, we demonstrate that IL-23 and IL-1 secreted by DCs in response to microbial stimulation work together to induce IL-17A production by ILCs. TNF is not required for IL-17A secretion by ILCs in vitro but synergizes with IL-17A to induce the expression of neutrophil-attracting chemokines. Upstream, activation of the IL-23/IL-17A axis is regulated by nucleotide-binding oligomerization domain containing (Nod)/receptor-interacting serine-threonine kinase 2 (Ripk2) signals in DCs. Genetic ablation of the Nod/Ripk2 signaling pathway protects TRUC mice from developing colitis without affecting the colitogenicity of the intestinal microbiota. Our data provide insight into the complex network of interactions between IL-17A-secreting ILCs and other components of the innate immune system in the development of colitis.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Khmaladze, I., et al (2014). "Mannan induces ROS-regulated, IL-17A-dependent psoriasis arthritis-like disease in mice" Proc Natl Acad Sci U S A 111(35): E3669-3678.
PubMed
Psoriasis (Ps) and psoriasis arthritis (PsA) are poorly understood common diseases, induced by unknown environmental factors, affecting skin and articular joints. A single i.p. exposure to mannan from Saccharomyces cerevisiae induced an acute inflammation in inbred mouse strains resembling human Ps and PsA-like disease, whereas multiple injections induced a relapsing disease. Exacerbation of disease severity was observed in mice deficient for generation of reactive oxygen species (ROS). Interestingly, restoration of ROS production, specifically in macrophages, ameliorated both skin and joint disease. Neutralization of IL-17A, mainly produced by gammadelta T cells, completely blocked disease symptoms. Furthermore, mice depleted of granulocytes were resistant to disease development. In contrast, certain acute inflammatory mediators (C5, Fcgamma receptor III, mast cells, and histamine) and adaptive immune players (alphabeta T and B cells) were redundant in disease induction. Hence, we propose that mannan-induced activation of macrophages leads to TNF-alpha secretion and stimulation of local gammadelta T cells secreting IL-17A. The combined action of activated macrophages and IL-17A produced in situ drives neutrophil infiltration in the epidermis and dermis of the skin, leading to disease manifestations. Thus, our finding suggests a new mechanism triggered by exposure to exogenous microbial components, such as mannan, that can induce and exacerbate Ps and PsA.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Schulze, F. S., et al (2014). "Fcgamma receptors III and IV mediate tissue destruction in a novel adult mouse model of bullous pemphigoid" Am J Pathol 184(8): 2185-2196.
PubMed
Bullous pemphigoid (BP) and epidermolysis bullosa acquisita are subepidermal autoimmune blistering diseases mediated by autoantibodies against type XVII collagen (Col17) and Col7, respectively. For blister formation, Fc-mediated events, such as infiltration of inflammatory cells in the skin, complement activation, and release of proteases at the dermal-epidermal junction, are essential. Although in the neonatal passive transfer mouse model of BP, tissue destruction is mediated by Fcgamma receptors (FcgammaRs) I and III, the passive transfer model of epidermolysis bullosa acquisita completely depends on FcgammaRIV. To clarify this discrepancy, we developed a novel experimental model for BP using adult mice. Lesion formation was Fc mediated because gamma-chain-deficient mice and mice treated with anti-Col17 IgG, depleted from its sugar moiety at the Fc portion, were resistant to disease induction. By the use of various FcgammaR-deficient mouse strains, tissue destruction was shown to be mediated by FcgammaRIV, FcgammaRIII, and FcgammaRIIB, whereas FcgammaRI was not essential. Furthermore, anti-inflammatory mediators in already clinically diseased mice can be explored in the novel BP model, because the pharmacological inhibition of FcgammaRIV and depletion of granulocytes abolished skin blisters. Herein, we extended our knowledge about the importance of FcgammaRs in experimental BP and established a novel BP mouse model suitable to study disease development over a longer time period and explore novel treatment strategies in a quasi-therapeutic setting.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
Norris, B. A., et al (2013). "Chronic but not acute virus infection induces sustained expansion of myeloid suppressor cell numbers that inhibit viral-specific T cell immunity" Immunity 38(2): 309-321.
PubMed
Resolution of acute and chronic viral infections requires activation of innate cells to initiate and maintain adaptive immune responses. Here we report that infection with acute Armstrong (ARM) or chronic Clone 13 (C13) strains of lymphocytic choriomeningitis virus (LCMV) led to two distinct phases of innate immune response. During the first 72 hr of infection, dendritic cells upregulated activation markers and stimulated antiviral CD8(+) T cells, independent of viral strain. Seven days after infection, there was an increase in Ly6C(hi) monocytic and Gr-1(hi) neutrophilic cells in lymphoid organs and blood. This expansion in cell numbers was enhanced and sustained in C13 infection, whereas it occurred only transiently with ARM infection. These cells resembled myeloid-derived suppressor cells and potently suppressed T cell proliferation. The reduction of monocytic cells in Ccr2(-/-) mice or after Gr-1 antibody depletion enhanced antiviral T cell function. Thus, innate cells have an important immunomodulatory role throughout chronic infection.
in vivo depletion of Gr-1+ myeloid cells
Flow Cytometry
van der Merwe, M., et al (2013). "Recipient myeloid-derived immunomodulatory cells induce PD-1 ligand-dependent donor CD4+Foxp3+ regulatory T cell proliferation and donor-recipient immune tolerance after murine nonmyeloablative bone marrow transplant
PubMed
We showed previously that nonmyeloablative total lymphoid irradiation/rabbit anti-thymocyte serum (TLI/ATS) conditioning facilitates potent donor-recipient immune tolerance following bone marrow transplantation (BMT) across MHC barriers via recipient invariant NKT (iNKT) cell-derived IL-4-dependent expansion of donor Foxp3(+) naturally occurring regulatory T cells (nTregs). In this study, we report a more specific mechanism. Wild-type (WT) BALB/c (H-2(d)) hosts were administered TLI/ATS and BMT from WT or STAT6(-/-) C57BL/6 (H-2(b)) donors. Following STAT6(-/-) BMT, donor nTregs demonstrated no loss of proliferation in vivo, indicating that an IL-4-responsive population in the recipient, rather than the donor, drives donor nTreg proliferation. In graft-versus-host disease (GVHD) target organs, three recipient CD11b(+) cell subsets (Gr-1(high)CD11c(-), Gr-1(int)CD11c(-), and Gr-1(low)CD11c(+)) were enriched early after TLI/ATS + BMT versus total body irradiation/ATS + BMT. Gr-1(low)CD11c(+) cells induced potent H-2K(b+)CD4(+)Foxp3(+) nTreg proliferation in vitro in 72-h MLRs. Gr-1(low)CD11c(+) cells were reduced significantly in STAT6(-/-) and iNKT cell-deficient Jalpha18(-/-) BALB/c recipients after TLI/ATS + BMT. Depletion of CD11b(+) cells resulted in severe acute GVHD, and adoptive transfer of WT Gr-1(low)CD11c(+) cells to Jalpha18(-/-) BALB/c recipients of TLI/ATS + BMT restored day-6 donor Foxp3(+) nTreg proliferation and protection from CD8 effector T cell-mediated GVHD. Blockade of programmed death ligand 1 and 2, but not CD40, TGF-beta signaling, arginase 1, or iNOS, inhibited nTreg proliferation in cocultures of recipient-derived Gr-1(low)CD11c(+) cells with donor nTregs. Through iNKT-dependent Th2 polarization, myeloid-derived immunomodulatory dendritic cells are expanded after nonmyeloablative TLI/ATS conditioning and allogeneic BMT, induce PD-1 ligand-dependent donor nTreg proliferation, and maintain potent graft-versus-host immune tolerance.
in vivo depletion of Gr-1+ myeloid cells
Ordonez-Rueda, D., et al (2012). "A hypomorphic mutation in the Gfi1 transcriptional repressor results in a novel form of neutropenia" Eur J Immunol 42(9): 2395-2408.
PubMed
Using N-ethyl-N-nitrosourea-induced mutagenesis, we established a mouse model with a novel form of neutropenia resulting from a point mutation in the transcriptional repressor Growth Factor Independence 1 (Gfi1). These mice, called Genista, had normal viability and no weight loss, in contrast to mice expressing null alleles of the Gfi1 gene. Furthermore, the Genista mutation had a very limited impact on lymphopoiesis or on T- and B-cell function. Within the bone marrow (BM), the Genista mutation resulted in a slight increase of monopoiesis and in a block of terminal granulopoiesis. This block occurred just after the metamyelocytic stage and resulted in the generation of small numbers of atypical CD11b(+) Ly-6G(int) neutrophils, the nuclear morphology of which resembled that of mature WT neutrophils. Unexpectedly, once released from the BM, these atypical neutrophils contributed to induce mild forms of autoantibody-induced arthritis and of immune complex-mediated lung alveolitis. They additionally failed to provide resistance to acute bacterial infection. Our study demonstrates that a hypomorphic mutation in the Gfi1 transcriptional repressor results in a novel form of neutropenia characterized by a split pattern of functional responses, reflecting the distinct thresholds required for eliciting neutrophil-mediated inflammatory and anti-infectious responses.
in vivo depletion of Gr-1+ myeloid cells
Carr, K. D., et al (2011). "Specific depletion reveals a novel role for neutrophil-mediated protection in the liver during Listeria monocytogenes infection" Eur J Immunol 41(9): 2666-2676.
PubMed
Previous studies have suggested that neutrophils are required for resistance during infection with multiple pathogenic microorganisms. However, the depleting antibody used in those studies binds to both Ly6G and Ly6C (anti-Gr-1; clone RB6-8C5). This antibody has been shown to deplete not only neutrophils but also monocytes and a subset of CD8(+) T cells. Recently, an antibody against Ly6G, which specifically depletes neutrophils, was characterized. In the present study, neutrophils are depleted using the antibody against Ly6G during infection with the intracellular bacterium Listeria monocytogenes (LM). Our data show that neutrophil-depleted mice are much less susceptible to infection than mice depleted with anti-Gr-1. Although neutrophils are required for clearance of LM, their importance is more pronounced in the liver and during a high-dose bacterial challenge. Furthermore, we demonstrate that the protection mediated by neutrophils is due to the production of TNF-alpha, but not IFN-gamma. Additionally, neutrophils are not required for the recruitment of monocytes or the generation of adaptive T-cell responses during LM infection. This study highlights the importance of neutrophils during LM infection, and indicate that depletion of neutrophils is less detrimental to the host than depletion of all Gr-1-expressing cell populations.
in vivo depletion of Gr-1+ myeloid cells
Waight, J. D., et al (2011). "Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism" PLoS One 6(11): e27690.
PubMed
Myeloid-derived suppressor cells (MDSC) are induced under diverse pathologic conditions, including neoplasia, and suppress innate and adaptive immunity. While the mechanisms by which MDSC mediate immunosuppression are well-characterized, details on how they develop remain less understood. This is complicated further by the fact that MDSC comprise multiple myeloid cell types, namely monocytes and granulocytes, reflecting diverse stages of differentiation and the proportion of these subpopulations vary among different neoplastic models. Thus, it is thought that the type and quantities of inflammatory mediators generated during neoplasia dictate the composition of the resultant MDSC response. Although much interest has been devoted to monocytic MDSC biology, a fundamental gap remains in our understanding of the derivation of granulocytic MDSC. In settings of heightened granulocytic MDSC responses, we hypothesized that inappropriate production of G-CSF is a key initiator of granulocytic MDSC accumulation. We observed abundant amounts of G-CSF in vivo, which correlated with robust granulocytic MDSC responses in multiple tumor models. Using G-CSF loss- and gain-of-function approaches, we demonstrated for the first time that: 1) abrogating G-CSF production significantly diminished granulocytic MDSC accumulation and tumor growth; 2) ectopically over-expressing G-CSF in G-CSF-negative tumors significantly augmented granulocytic MDSC accumulation and tumor growth; and 3) treatment of naive healthy mice with recombinant G-CSF protein elicited granulocytic-like MDSC remarkably similar to those induced under tumor-bearing conditions. Collectively, we demonstrated that tumor-derived G-CSF enhances tumor growth through granulocytic MDSC-dependent mechanisms. These findings provide us with novel insights into MDSC subset development and potentially new biomarkers or targets for cancer therapy.
Immunohistochemistry (paraffin)
Li, M., et al (2006). "Topical vitamin D3 and low-calcemic analogs induce thymic stromal lymphopoietin in mouse keratinocytes and trigger an atopic dermatitis" Proc Natl Acad Sci U S A 103(31): 11736-11741.
PubMed
We have demonstrated that cytokine thymic stromal lymphopoietin (TSLP), whose expression is rapidly induced upon keratinocyte-selective ablation of retinoid X receptors (RXRs) -alpha and -beta in the mouse (RXRalphabeta(ep-/-) mice), plays a key role in initiating a skin and systemic atopic dermatitis-like phenotype. We show here that topical application of the physiologically active ligand [1alpha,25-(OH)(2)D(3); calcitriol] of the vitamin D receptor, or of its low-calcemic analog MC903 (calcipotriol; Dovonex), induces TSLP expression in epidermal keratinocytes, which results in an atopic dermatitis-like syndrome mimicking that seen in RXRalphabeta(ep-/-) mutants and transgenic mice overexpressing TSLP in keratinocytes. Furthermore, topical application of retinoic acid receptor RARgamma-selective agonist BMS961 also induces TSLP expression either on its own or synergistically with 1alpha,25-(OH)(2)D(3). Our data demonstrate that RXR/vitamin D receptor and RXR/retinoic acid receptor-gamma heterodimers and their ligands cell-autonomously control the expression of TSLP in epidermal keratinocytes of the mouse. We propose molecular mechanisms through which vitamin D3 and retinoic acid signalings could be involved in the pathogenesis of atopic diseases.
Immunohistochemistry (frozen)
Brown, C. R., et al (2004). "Treatment of mice with the neutrophil-depleting antibody RB6-8C5 results in early development of experimental lyme arthritis via the recruitment of Gr-1- polymorphonuclear leukocyte-like cells" Infect Immun 72(9): 4956-49
PubMed
Recently, we demonstrated that blocking the entry of neutrophils into Borrelia burgdorferi-infected joints in mice deficient in the chemokine receptor CXCR2 prevented the development of experimental Lyme arthritis. Neutrophils were marginalized in blood vessels at the site of infection but could not enter the joint tissue. In the present study, we treated both genetically arthritis-resistant DBA/2J (DBA) and arthritis-susceptible C3H/HeJ (C3H) mice with the neutrophil-depleting monoclonal antibody RB6-8C5 (RB6) to determine the effect on arthritis development. Surprisingly, both DBA and C3H mice treated with RB6 developed arthritis at 1 week postinfection, approximately 1 week earlier than the control-treated C3H mice. The early development of arthritis in the RB6-treated mice was accompanied by an influx into the joints of cells with ring-shaped polymorphonuclear leukocyte (PMN) cell morphology that were negative for the Gr-1 neutrophil maturation marker. RB6 treatment of mice also resulted in increased numbers of B. burgdorferi cells in the joints at 7 days postinfection and earlier expression of the chemokines KC and monocyte chemoattractant protein 1 in the joints compared to control-treated animals. Together, these results suggest that recruitment of neutrophils or PMN-like cells into an infected joint is a key requirement for Lyme arthritis development and that altered recruitment of these cells into the joints of arthritis-resistant mice can exacerbate the development of pathology.
Product Citations
-
-
Immunology and Microbiology
-
Cancer Research
Escherichia coli promotes colorectal cancer metastasis by maintaining enhancer-promoter loops through releasing neutrophil extracellular traps.
In Nat Commun on 3 February 2026 by Pan, B., Yao, Y., et al.
PubMed
The involvement of intestinal microbiota in the process of neutrophil-mediated colorectal cancer liver metastasis (CRCLM) is not yet fully understood. Here, we show that Escherichia coli (E. coli) is prevalent in CRC tissues with LM using 2bRAD-M-Seq and is linked to the release of neutrophil extracellular traps (NETs). Utilizing multi-omics and molecular techniques, we establish that E. coli recruits RIPK2, which promotes the binding of HNRNPK to the Atf3/Relb promoters in neutrophils, thereby enhancing their transcription. This process results in the upregulation of Ncf4, which triggers p-MLKL-mediated NET formation. NETs, in turn, increase the expression of TRPC1 and NFATC3 in CRC cells, promoting the calcium-dependent assembly of the STAT3/S100A8/9 heterotrimer. This trimer stabilizes STAT3-enhancer-promoter loops (EPLs), thereby reinforcing the Tns1 transcription and facilitating CRCLM. Our findings elucidate the mechanism by which E. coli-induced NETs promote CRCLM through epigenetic modifications, offering an insight into the role of EPLs in immune regulation and tumor progression.
-
-
-
Biochemistry and Molecular biology
-
Cell Biology
Control of renal central carbon metabolism by heme oxygenase-1.
In iScience on 16 January 2026 by Guerra, J., Jentho, E., et al.
PubMed
Metabolic adaptation is an integral part of the organismal stress-response. In this study, we investigate the role of Hmox1 in mediating metabolic adaptation under both physiological and hemolytic stress conditions. Using an inducible Hmox1 deletion model (Hmox1R26Δ/Δ ), we demonstrate that Hmox1 expression is essential for preventing heme-induced kidney failure. Our integrative approach, combining bulk RNA sequencing and targeted metabolomics within silico metabolic modeling, revealed a compromised pentose phosphate pathway (PPP) in failed renal adaptation after Hmox1 deletion. This study underscores the critical role of Hmox1 for PPP regulation and sheds light on the metabolic pathways involved in kidney dysfunction due to hemolytic diseases.
-
-
-
Immunology and Microbiology
-
Cancer Research
IL-17-producing γδ T cells in the tumor microenvironment promote radioresistance in mice.
In J Clin Invest on 15 December 2025 by Deng, Y., Liu, X., et al.
PubMed
The immunosuppressive tumor microenvironment (TME) drives radioresistance, but the role of γδ T cells in regulating radiosensitivity remains incompletely understood. In this study, we found that γδ T cell infiltration in the TME substantially increased after radiotherapy and contributed to radioresistance. Depletion of γδ T cells enhanced radiosensitivity. Single-cell RNA-seq revealed that γδ T cells in the postradiotherapy TME were characterized by the expression of Zbtb16, Il23r, and Il17a, and served as the primary source of IL-17A. These γδ T cells promoted radioresistance by recruiting myeloid-derived suppressor cells and suppressing T cell activation. Mechanistically, radiotherapy-induced tumor cell-derived microparticles containing dsDNA activated the cGAS-STING/NF-κB signaling pathway in macrophages, upregulating the expression of the chemokine CCL20, which was critical for γδ T cell recruitment. Targeting γδ T cells and IL-17A enhanced radiosensitivity and improved the efficacy of radiotherapy combined with anti-PD-1 immunotherapy, providing potential therapeutic strategies to overcome radioresistance.
-
-
-
Immunology and Microbiology
-
Cancer Research
Targeting MDA5 enhances tumor immunity through immunogenic cell death and augments the efficacy of immune checkpoint blockade
In Research Square on 9 December 2025 by Wang, W., Niu, Z., et al.
-
-
-
Biochemistry and Molecular biology
-
Immunology and Microbiology
CARD14 signaling in intestinal epithelial cells induces intestinal inflammation and intestinal transit delay.
In EMBO Mol Med on 1 December 2025 by Aidarova, A., Carels, M., et al.
PubMed
CARD14 is an intracellular NF-κB signaling mediator in the skin, and rare CARD14 variants have been associated with psoriasis and atopic dermatitis. CARD14 is also expressed in intestinal epithelial cells (IEC). However, its function in the intestine remains unknown. We demonstrate here that transgenic mice expressing the psoriasis-associated gain-of-function human CARD14(E138A) mutant specifically in IEC show mild intestinal inflammation, without epithelial damage. Moreover, CARD14(E138A)IEC mice show a drastic reduction in intestinal motility, often associated with rectal prolapse. Enteric neuronal survival and functionality are unaffected in CARD14(E138A)IEC mice. Transcriptome analysis of IEC from CARD14(E138A)IEC mice reveals decreased expression of antimicrobial peptides by Paneth cells, accompanied by microbial dysbiosis and increased susceptibility to enteric bacterial infection. Our findings suggest that gain-of-function CARD14 mutations may not only predispose patients to psoriasis but also mild intestinal inflammation, reduced intestinal motility, and increased sensitivity to intestinal infection. CARD14(E138A)IEC mice are also a valuable tool for further investigation of IEC-intrinsic molecular processes involved in intestinal inflammation and motility disorders.
-
-
-
Cancer Research
-
Immunology and Microbiology
TGFβ signaling in cancer-associated fibroblasts drives a hepatic gp130-dependent pro-metastatic inflammatory program in colorectal cancer.
In iScience on 21 November 2025 by Harryvan, T. J., Abudukelimu, S., et al.
PubMed
The Consensus Molecular Subtype 4 (CMS4) of colorectal cancer (CRC) has the worst prognosis and the highest frequency of hepatic metastases. It is characterized by abundant cancer-associated fibroblasts (CAFs) in the tumor microenvironment and active TGFβ signaling, but the molecular drivers of metastasis remain unclear. Here, we show that TGFβ signaling in CRC patient-derived CAFs from the primary tumor induces production of IL-6 family cytokines, particularly IL-6 and IL-11. These cytokines stimulate hepatocytes to express myeloid chemoattractants, including SAA1, through gp130-dependent JAK/STAT signaling. This promotes neutrophil recruitment to the liver, potentially creating a pro-metastatic niche. This IL-6 family-JAK/STAT stromal signaling axis is active in both a murine model of CMS4 as well as in patients with human CRC in vivo. Combined, our data reveal that TGFβ-driven CAF signaling actively contributes to the formation of a neutrophil-dependent, pre-metastatic hepatic niche in the metastatic phenotype of CMS4 CRC.
-
-
Mucoricin binding to β-glucan sites on germinating Mucorales spores disrupts neutrophil swarming to promote pathogenicity
In bioRxiv on 29 October 2025 by Baimpa, S., Sertedakis, M., et al.
-
Neutrophil extracellular traps-mediated thrombosis drive pyrrolizidine alkaloid-induced hepatic sinusoidal obstruction syndrome
In bioRxiv on 22 October 2025 by Shuang, Z., Dongming, Y., et al.
-
-
Pharmacology
FGF1ΔHBS ameliorates DSS-induced ulcerative colitis by reducing neutrophil recruitment through the MAPK pathway.
In Br J Pharmacol on 1 October 2025 by Feng, S., Jin, Y., et al.
PubMed
Inflammatory bowel diseases (IBDs) constitute chronic inflammatory disease of the gastrointestinal tract, with escalating global prevalence. There is a pressing demand for safe and effective treatments for IBDs. Fibroblast growth factor 1 (FGF1) variant FGF1ΔHBS, characterised by reduced mitogenic capacity, has shown promising therapeutic potential in various inflammatory conditions, including obesity and diabetic nephropathy. Hence, exploring the therapeutic impact of FGF1ΔHBS on colitis is warranted.
-
-
-
Cancer Research
Highly Selective Exogenous Neutrophils Effectively Inhibit Growth of Colon Cancer under the Guidance of Precision Navigation.
In Research (Wash D C) on 18 September 2025 by Yang, Y., Li, C., et al.
PubMed
The diversity of neutrophils' subtypes and functions in the tumor microenvironmentunderlies their contributions to both pro- and anti-tumorigenic activities. How to elucidate the antitumor mechanisms of neutrophil and effectively utilize it still needs to be studied. In this study, neutrophil stimulus reaction index (NSRI) was established for high selectivity of neutrophils. Sufficient and highly selective exogenous neutrophils were used to infuse into tumor-bearing mice. Navigation technique was used to make neutrophils rapidly infiltrate into the tumor tissue, which played a key role in inhibiting tumor growth. We found that tumor cells were efficiently killed in direct coculture with sufficient neutrophils by promoting a multimodal death primarily dominated by apoptosis. Interestingly, colon cancer organoid diameters were decreased markedly cocultured with neutrophils, and were more remarkable in the application of organoid microinjection of neutrophils. Consistent with the 3D-printed model, in vivo model indicated that neutrophils meaningfully inhibited the tumor growth by formation of neutrophil extracellular traps (NETs) with neutrophil elastase (NE) payload. This study not only unveils the crucial anticancer effects of neutrophils but also sets out an innovative strategy of aggressive dose exogenous neutrophils with precise navigation, which provided the basis for advancements in neutrophil-based immunotherapy for colon cancer treatment.
-
-
-
Cancer Research
-
Immunology and Microbiology
Hepatocyte-Derived LRG1 Primes the Liver for Metastasis and Impairs Immunotherapy
In Research Square on 17 September 2025 by Wang, W., Long, G., et al.
-
-
Chronic social defeat stress induces meningeal neutrophilia via type I interferon signaling in male mice.
In Nat Commun on 1 September 2025 by Kigar, S. L., Lynall, M. E., et al.
PubMed
Inflammation is increasingly recognized as a risk factor for psychiatric disorders. Animal models of stress and stress-related disorders are associated with blood neutrophilia. The mechanistic relevance of this to symptoms or behavior is unclear. We characterized the immune response to chronic social defeat (CSD) stress at brain border regions in male mice. Here we show that chronic, but not acute, stress causes neutrophil accumulation in the meninges-i.e., "meningeal neutrophilia"- but not the brain. CSD promotes neutrophil trafficking to meninges via vascular channels originating from skull bone marrow (BM). Transcriptional analysis suggests CSD increases type I interferon (IFN-I) signaling in meningeal neutrophils. Blocking this pathway via the IFN-I receptor (IFNAR) protects against the negative behavioral effects of CSD stress. Our identification of IFN-I signaling as a putative mediator of meningeal neutrophil recruitment may facilitlate development of new therapies for stress-related disorders.
-
-
Cancer Research
Liver regeneration-associated hepatocellular YAP1 activation prevents colorectal cancer liver metastasis through glutamine competition.
In Sci Adv on 22 August 2025 by Yu, Q., Yu, M., et al.
PubMed
The literature suggests that hepatocellular Yes-associated protein 1 (YAP1) signaling is activated following hepatectomy and that such activation can suppress the growth of metastatic liver tumors. The prognosis of a real-world cohort of 240 patients with colorectal cancer liver metastasis (CRLM) undergoing major and minor hepatectomy was compared after adjusting for confounding factors. To model CRLM, we induced liver metastasis in mice by transsplenically injecting MC38 cells. We found that patients with CRLM and mice undergoing major hepatectomy had better survival compared to those undergoing minor hepatectomy. Mechanistically, extensive hepatectomy activates hepatocellular YAP1 by regulating the epidermal growth factor receptor, altering glutamine metabolism-related gene expression and increasing liver glutamine consumption. This metabolic shift leads to glutamine scarcity in tumor cells, causing increased reactive oxygen species production, which promotes loss of YAP1 activity in tumor cells. Consequently, the production of the chemokine CXCL5 is suppressed, which inhibits myeloid-derived suppressor cell infiltration and enhancing the immunological function of CD8+ T cells.
-
-
-
Neuroscience
-
Immunology and Microbiology
-
Cardiovascular biology
Microglia are prominent producers of inflammatory cytokines during the hyperacute phase of ischemic stroke.
In Commun Biol on 10 August 2025 by Bourne, J. H., Suthya, A. R., et al.
PubMed
Current treatments for ischemic stroke focus on removing neurovascular-occluding clots but overlook the resulting neuroinflammation. Clinical trials targeting brain-infiltrating peripheral immune cells have failed to improved outcomes, leaving mechanisms of neuroinflammation poorly understood. Whilst many studies have examined the inflammatory cells and cytokine profile of the post-stroke brain at days and weeks following injury onset, we propose that interventions at these timepoints are too late to limit brain damage. In this study, we examined brain immune cell composition, cytokine levels, and neurological dysfunction at hyperacute (3 h) and acute (24 h) stages following a preclinical mouse model of ischemic stroke. Interestingly, we detected elevated inflammatory cytokines in brain tissue as early as 3 h, notably before infiltrating neutrophil and monocyte arrival at 24 h. Depletion of peripheral immune cells by antibodies or genetic alteration did not dampen neuroinflammation, nor improve sensorimotor function. Intravital imaging of Cx3cr1gfp/+ mice showed that microglia, the brain-resident immune cell, display rapidly altered morphology, and swiftly upregulated cytokine production hyperacutely post-stroke. Altogether, our study provides evidence that microglia are key drivers of early neuroinflammation and modulating their function at clinically-relevant timepoints will improve stroke recovery.
-
-
-
Immunology and Microbiology
Characterisation of an autochthonous mouse ccRCC model of immune checkpoint inhibitor therapy resistance.
In Sci Rep on 5 June 2025 by Peighambari, A., Huang, H., et al.
PubMed
Many metastatic clear cell renal cell carcinomas (ccRCC) are resistant to immune checkpoint inhibitor therapies, however the mechanisms underlying sensitivity or resistance remain incompletely characterised. We demonstrate that ccRCCs in the Vhl/Trp53/Rb1 mutant mouse model are resistant to combined anti-PD-1/anti-CTLA-4 therapy alone and in combination with additional therapeutic agents that reflect current ccRCC clinical trials. However, in some animals in vivo checkpoint therapy allowed isolated splenic T cells to recognise cultured ccRCC cells from the same animal, implicating the tumour microenvironment in suppression of T cell activation. We identified putative immunosuppressive myeloid cell populations with features similar to myeloid cells in the microenvironment of human ccRCC. The expression patterns of immune checkpoint ligands in both the mouse model and in human ccRCC suggests that several checkpoint systems other than PD-1 and CTLA-4 are likely to represent the dominant T cell suppressive forces in ccRCC. Our findings characterise an autochthonous mouse ccRCC model of immune checkpoint inhibitor therapy resistance and pave the way for a systematic functional dissection of the identified potential molecular barriers to effective immune therapy of ccRCC.
-
-
-
Cardiovascular biology
Acute kidney injury triggers hypoxemia by lung intravascular neutrophil retention that reduces capillary blood flow.
In J Clin Invest on 15 May 2025 by Komaru, Y., Ning, L., et al.
PubMed
Sterile acute kidney injury (AKI) is common in the clinic and frequently associated with unexplained hypoxemia that does not improve with dialysis. AKI induces remote lung inflammation with neutrophil recruitment in mice and humans, but which cellular cues establish neutrophilic inflammation and how it contributes to hypoxemia is not known. Here we report that AKI induced rapid intravascular neutrophil retention in lung alveolar capillaries without extravasation into tissue or alveoli, causing hypoxemia by reducing lung capillary blood flow in the absence of substantial lung interstitial or alveolar edema. In contrast to direct ischemic lung injury, lung neutrophil recruitment during remote lung inflammation did not require cues from intravascular nonclassical monocytes or tissue-resident alveolar macrophages. Instead, lung neutrophil retention depended on the neutrophil chemoattractant CXCL2 released by activated classical monocytes. Comparative single-cell RNA-Seq analysis of direct and remote lung inflammation revealed that alveolar macrophages were highly activated and produced CXCL2 only in direct lung inflammation. Establishing a CXCL2 gradient into the alveolus by intratracheal CXCL2 administration during AKI-induced remote lung inflammation enabled neutrophils to extravasate. We thus discovered important differences in lung neutrophil recruitment in direct versus remote lung inflammation and identified lung capillary neutrophil retention that negatively affected oxygenation by causing a ventilation-perfusion mismatch as a driver of AKI-induced hypoxemia.
-
-
-
Immunology and Microbiology
Mac-1 regulates disease stage-specific immunosuppression via the nitric oxide pathway in autoimmune disease.
In Sci Adv on 9 May 2025 by Wang, W., Cao, C., et al.
PubMed
Integrin Mac-1 plays a critical role in the development of multiple sclerosis (MS); however, the underlying mechanism is not fully understood. Here, we developed a myeloid-specific Mac-1-deficient mouse. Using an experimental autoimmune encephalomyelitis (EAE) mouse model of MS, we report that Mac-1 on myeloid cells is key to disease development. Our data reveal that myeloid-specific Mac-1 significantly increases EAE severity and hinders disease regression. Loss of Mac-1 increases Gr-1+ cells in peripheral tissues and the CNS and preferably accelerates the transition of Ly6Chi monocytes from a pro-inflammatory to an immunosuppressive phenotype in a disease stage-dependent manner. Mechanistically, our results demonstrate that Mac-1 suppresses interferon-γ production and prevents monocytes from acquiring immunosuppressive functions by reducing the expression of iNOS, IDO, and CD84. Administration of a NOS-specific inhibitor in Mac-1-deficient EAE mice abolishes disease regression. These insights could help develop Mac-1-targeting strategies for better treatment of MS.
-
-
Selective STING Activation in Intratumoral Myeloid Cells via CCR2-Directed Antibody-Drug Conjugate TAK-500.
In Cancer Immunol Res on 2 May 2025 by Appleman, V. A., Matsuda, A., et al.
PubMed
The tumor microenvironment in solid tumors contains myeloid cells that modulate local immune activity. Stimulator of IFN gene (STING) signaling activation in these myeloid cells enhances local type-I IFN production, inducing an innate immune response that mobilizes adaptive immunity and reprograms immunosuppressive myeloid populations to drive antitumor immunity. In this study, we generated TAK-500, an immune cell-directed antibody-drug conjugate, to deliver a STING agonist to CCR2+ human cells and drive enhanced antitumor activity relative to nontargeted STING agonists. Preclinically, TAK-500 triggered dose-dependent innate immune activation in vitro. In addition, a murine TAK-500 immune cell-directed antibody-drug conjugate surrogate enhanced innate and adaptive immune responses both in in vitro and murine tumor models. Spatially resolved analysis of CCR2 and immune cell markers in the tumor microenvironment of >1,000 primary human tumors showed that the CCR2 protein was predominantly expressed in intratumoral myeloid cells. Collectively, these data highlight the clinical potential of delivering a STING agonist to CCR2+ cells.
-
-
Immunology and Microbiology
Interleukin-35 inhibits NETs to ameliorate Th17/Treg immune imbalance during the exacerbation of cigarette smoke exposed-asthma via gp130/STAT3/ferroptosis axis.
In Redox Biol on 1 May 2025 by Tao, P., Su, B., et al.
PubMed
Cigarette smoke (CS) exposure amplifies neutrophil accumulation. IL-35, a novel cytokine with anti-inflammatory properties, is involved in protection against asthma. However, the biological roles of neutrophils and the precise molecular mechanisms of IL-35 in CS exposed-asthma remain unclear. We showed that the exacerbation of CS exposed-asthma leads to dramatically increased neutrophil counts and an imbalance in DC-Th17/Treg immune responses. RNA sequencing revealed that NETs, part of a key biological process in neutrophils, were significantly upregulated in the context of CS exposed-asthma exacerbation and that IL-35 treatment downregulated NET-associated gene expression. Targeted degradation of NETs, rather than neutrophil depletion, alleviated the CS exposed-asthma. Mechanistically, STAT3 phosphorylation promoted ferroptosis, exacerbating NET release, which in turn enhanced dendritic cell (DC) antigen presentation, activated T cells, and specifically promoted Th17 cell differentiation while inhibiting Treg cells. IL-35 acting on the gp130 receptor alleviated STAT3-mediated ferroptosis-associated NET formation. In summary, our study revealed a novel mechanism by which IL-35 inhibited NET formation, subsequently alleviating neutrophilic inflammation and restoring the DC-Th17/Treg imbalance in CS exposed-asthma, highlighting the potential of IL-35 as a targeted therapeutic strategy.
-
-
-
Cancer Research
METTL3 promotes an immunosuppressive microenvironment in bladder cancer via m6A-dependent CXCL5/CCL5 regulation.
In J Immunother Cancer on 15 April 2025 by Tong, Y., Chen, Z., et al.
PubMed
Bladder cancer (BLCA) is a challenging malignancy with a poor prognosis, particularly in muscle-invasive cases. Despite recent advancements in immunotherapy, response rates remain suboptimal. This study investigates the role of METTL3, an m6A RNA methylation "writer," in regulating the immune microenvironment of BLCA.
-