InVivoMAb anti-mouse CD8α
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
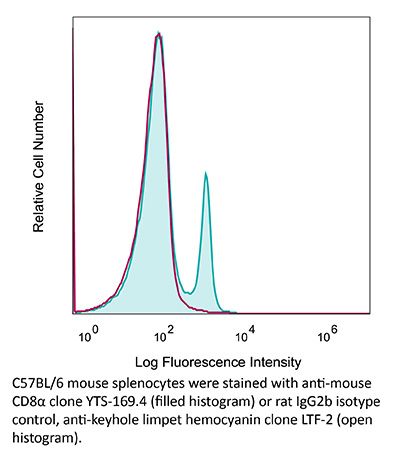
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | CBA mouse thymocytes |
| Reported Applications |
in vivo CD8+ T cell depletion Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_10950145 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Li, Z., et al (2015). "Pre-treatment of allogeneic bone marrow recipients with the CXCR4 antagonist AMD3100 transiently enhances hematopoietic chimerism without promoting donor-specific skin allograft tolerance" Transpl Immunol 33(2): 125-129.
PubMed
Hematopoietic chimerism established by allogeneic bone marrow transplantation is known to promote donor-specific organ allograft tolerance; however, clinical application is limited by the need for toxic host conditioning and “megadoses” of donor bone marrow cells. A potential solution to this problem has been suggested by the observation that recipient bone marrow mobilization by the CXCR4 antagonist AMD3100 promotes chimerism in congenic bone marrow transplantation experiments in mice. Here we report that a single subcutaneous dose of 10mg/kg AMD3100 in recipient C57BL/6 mice was able to enhance hematopoietic chimerism when complete MHC-mismatched BALB/c donor bone marrow cells were transplanted 1h after drug dosing. However, levels of chimerism measured 30days post-transplantation were not sustained when mice were reexamined on day 90 post-transplantation. Moreover, transient chimerism induced by this protocol did not support robust donor-specific skin allograft tolerance. Using the same transient immunosuppression protocol, we confirmed that “megadoses” of donor bone marrow cells could induce durable chimerism associated with donor-specific skin allograft tolerance without AMD3100 pre-treatment. We conclude that in this protocol AMD3100 pretreatment may empty bone marrow niches that become reoccupied by allogeneic donor hematopoietic progenitor cells but not by true long-lived donor hematopoietic stem cells, resulting in short-lived chimerism and failure to support durable donor-specific allograft tolerance.
-
Li, Z., et al (2015). "Pre-treatment of allogeneic bone marrow recipients with the CXCR4 antagonist AMD3100 transiently enhances hematopoietic chimerism without promoting donor-specific skin allograft tolerance" Transpl Immunol 33(2): 125-129.
PubMed
Hematopoietic chimerism established by allogeneic bone marrow transplantation is known to promote donor-specific organ allograft tolerance; however, clinical application is limited by the need for toxic host conditioning and “megadoses” of donor bone marrow cells. A potential solution to this problem has been suggested by the observation that recipient bone marrow mobilization by the CXCR4 antagonist AMD3100 promotes chimerism in congenic bone marrow transplantation experiments in mice. Here we report that a single subcutaneous dose of 10mg/kg AMD3100 in recipient C57BL/6 mice was able to enhance hematopoietic chimerism when complete MHC-mismatched BALB/c donor bone marrow cells were transplanted 1h after drug dosing. However, levels of chimerism measured 30days post-transplantation were not sustained when mice were reexamined on day 90 post-transplantation. Moreover, transient chimerism induced by this protocol did not support robust donor-specific skin allograft tolerance. Using the same transient immunosuppression protocol, we confirmed that “megadoses” of donor bone marrow cells could induce durable chimerism associated with donor-specific skin allograft tolerance without AMD3100 pre-treatment. We conclude that in this protocol AMD3100 pretreatment may empty bone marrow niches that become reoccupied by allogeneic donor hematopoietic progenitor cells but not by true long-lived donor hematopoietic stem cells, resulting in short-lived chimerism and failure to support durable donor-specific allograft tolerance.
-
Dai, M., et al (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257.
PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
-
Dai, M., et al (2013). "Long-lasting complete regression of established mouse tumors by counteracting Th2 inflammation" J Immunother 36(4): 248-257.
PubMed
40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.”}” data-sheets-userformat=”{“2″:14851,”3”:{“1″:0},”4”:{“1″:2,”2″:16777215},”12″:0,”14”:{“1″:2,”2″:1521491},”15″:”Roboto, sans-serif”,”16″:12}”>Mice with intraperitoneal ID8 ovarian carcinoma or subcutaneous SW1 melanoma were injected with monoclonal antibodies (mAbs) to CD137PD-1CTLA4 7-15 days after tumor initiation. Survival of mice with ID8 tumors tripled and >40% of mice with SW1 tumors remained healthy >150 days after last treatment and are probably cured. Therapeutic efficacy was associated with a systemic immune response with memory and antigen specificity, required CD4 cells and involved CD8 cells and NK cells to a less extent. The 3 mAb combination significantly decreased CD19 cells at tumor sites, increased IFN-gamma and TNF-alpha producing CD4 and CD8 T cells and mature CD86 dendritic cells (DC), and it increased the ratios of effector CD4 and CD8 T cells to CD4Foxp3 regulatory T (Treg) cells and to CD11bGr-1 myeloid suppressor cells (MDSC). This is consistent with shifting the tumor microenvironment from an immunosuppressive Th2 to an immunostimulatory Th1 type and is further supported by PCR data. Adding an anti-CD19 mAb to the 3 mAb combination in the SW1 model further increased therapeutic efficacy. Data from ongoing experiments show that intratumoral injection of a combination of mAbs to CD137PD-1CTLA4CD19 can induce complete regression and dramatically prolong survival also in the TC1 carcinoma and B16 melanoma models, suggesting that the approach has general validity.
Product Citations
-
Disruption of tRNA threonylation triggers RIG-I mediated anti-tumour immune response.
In Nat Commun on 25 February 2026 by Dziagwa, C., Seca, C., et al.
PubMed
Tumour-induced mechanisms of immune evasion hinder immune response to cancer, particularly in melanoma. mRNA translation, by ensuring accurate protein synthesis, regulates cancer phenotypes and immune response, but the underlying mechanisms remain unclear. Here, we reveal how O-sialoglycoprotein endopeptidase (OSGEP), catalysing the tRNA modification N6-threonylcarbamoyladenosine (t6A), drives protein homeostasis in cancer cells to maintain T-cell exclusion and prevent anti-tumour immune response. t6A-deficient melanoma cells disrupt efficient cytoplasmic translation of ANN codons (trinucleotides with A in the first position and N = any nucleotide), causing specific protein aggregation and the formation of integrated stress response-dependent stress granules. We discovered that OSGEP loss triggers melanoma regression by relocating RIG-I to stress granules, leading to its pathway activation. As a result, T-cells are recruited to the tumour site and orchestrate an anti-tumour immune response. Finally, an OSGEP-driven gene signature in melanoma patients is associated with T-cell infiltration and improved overall survival. Together, our findings position t6A tRNA modification as a promising therapeutic target for melanoma treatment.
-
CoREST complex inhibition alters RNA splicing to promote neoantigen expression and enhance tumor immunity.
In JCI Insight on 23 January 2026 by Fisher, R. J., Park, K., et al.
PubMed
Epigenetic macromolecular enzyme complexes tightly regulate gene expression at the chromatin level and have recently been found to colocalize with RNA splicing machinery during active transcription; however, the precise functional consequences of these interactions are uncertain. Here, we identify unique interactions of the CoREST repressor complex (LSD1-HDAC1-CoREST) with components of the RNA splicing machinery and their functional consequences in tumorigenesis. Using mass spectrometry, in vivo binding assays, and cryo-EM, we find that CoREST complex-splicing factor interactions are direct and perturbed by the CoREST complex selective inhibitor, corin, leading to extensive changes in RNA splicing in melanoma and other malignancies. Moreover, these corin-induced splicing changes are shown to promote global effects on oncogenic and survival-associated splice variants, leading to a tumor-suppressive phenotype. Using machine learning models, MHC IP-MS, and ELISpot assays, we identify thousands of neopeptides derived from unannotated splice sites that generate corin-induced splice-neoantigens that are demonstrated to be immunogenic in vitro. Corin is further shown to reactivate the response to immune checkpoint blockade, effectively sensitizing tumors to anti-PD-1 immunotherapy. These data position CoREST complex inhibition as a unique therapeutic opportunity that perturbs oncogenic splicing programs while also creating tumor-associated neoantigens that enhance the immunogenicity of current therapeutics.
-
RAS(ON) Multiselective Inhibition Drives Antitumor Immunity in Preclinical Models of NRAS-Mutant Melanoma.
In Cancer Immunol Res on 8 January 2026 by Carvalho, L. A., Tovbis Shifrin, N., et al.
PubMed
Targeted therapies for NRAS-mutant melanoma remain an unmet clinical need. In this study, we demonstrate that RMC-7977, a preclinical RAS(ON) multiselective inhibitor representative of the investigational agent daraxonrasib (RMC-6236), was able to elicit potent antitumor immune responses across multiple NRAS-mutant melanoma models. Treatment with RMC-7977 led to rapid tumor regressions driven by inhibition of MAPK signaling, upregulation of MHC and PD-L1 proteins, and enhanced infiltration of CD4+ and CD8+ T cells. Complete responses were dependent on adaptive immunity, as both CD4+ and CD8+ T cells were essential for extended survival. Resistance to treatment was marked by reduced T-cell infiltration, loss of MHC class I expression, and expansion of myeloid-derived suppressor cells. Combining RMC-7977 with anti-PD-1 boosted cytotoxic T-cell infiltration, reprogrammed myeloid cells toward an antigen-presenting phenotype, and improved survival in models resistant to PD-1 blockade. Consistent with these preclinical data, objective clinical responses were observed in two patients with NRAS-mutant melanoma treated with daraxonrasib in an ongoing phase I/Ib clinical trial. Together, these data support the continued clinical evaluation of RAS(ON) multiselective inhibitors for the treatment of NRAS-mutant melanoma.
-
Neoadjuvant personalized viral vaccine prevents tumor relapse in checkpoint-resistant murine melanoma model.
In J Immunother Cancer on 29 November 2025 by Seclì, L., Nocchi, L., et al.
PubMed
Personalized cancer vaccines targeting tumor-specific neoantigens (nAgs) are an emerging therapeutic strategy, particularly effective in early-stage disease before immune suppression is established. Immune checkpoint inhibitors have demonstrated benefit in the adjuvant setting (postsurgery), and recent evidence suggests neoadjuvant administration (before surgery) may further enhance antitumor immunity. This study evaluated the efficacy of a multiepitope nAg vaccine in a preclinical melanoma model resistant to checkpoint inhibition, comparing neoadjuvant and adjuvant treatment, alone or in combination with anti-programmed cell death protein 1 (PD1) therapy.