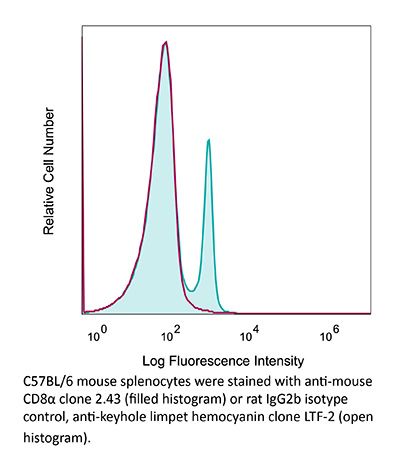
InVivoMAb anti-mouse CD8α
Product Description
Specifications
| Isotype | Rat IgG2b, κ |
|---|---|
| Recommended Isotype Control(s) | InVivoMAb rat IgG2b isotype control, anti-keyhole limpet hemocyanin |
| Recommended Dilution Buffer | InVivoPure pH 7.0 Dilution Buffer |
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Mouse CTL clone L3 |
| Reported Applications |
in vivo CD8+ T cell depletion Western blot |
| Formulation |
PBS, pH 7.0 Contains no stabilizers or preservatives |
| Endotoxin |
≤1EU/mg (≤0.001EU/μg) Determined by LAL assay |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1125541 |
| Molecular Weight | 150 kDa |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
-
Coffelt, S. B., et al (2015). "IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis" Nature 522(7556): 345-348.
PubMed
Metastatic disease remains the primary cause of death for patients with breast cancer. The different steps of the metastatic cascade rely on reciprocal interactions between cancer cells and their microenvironment. Within this local microenvironment and in distant organs, immune cells and their mediators are known to facilitate metastasis formation. However, the precise contribution of tumour-induced systemic inflammation to metastasis and the mechanisms regulating systemic inflammation are poorly understood. Here we show that tumours maximize their chance of metastasizing by evoking a systemic inflammatory cascade in mouse models of spontaneous breast cancer metastasis. We mechanistically demonstrate that interleukin (IL)-1beta elicits IL-17 expression from gamma delta (gammadelta) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils in mice bearing mammary tumours. Tumour-induced neutrophils acquire the ability to suppress cytotoxic T lymphocytes carrying the CD8 antigen, which limit the establishment of metastases. Neutralization of IL-17 or G-CSF and absence of gammadelta T cells prevents neutrophil accumulation and downregulates the T-cell-suppressive phenotype of neutrophils. Moreover, the absence of gammadelta T cells or neutrophils profoundly reduces pulmonary and lymph node metastases without influencing primary tumour progression. Our data indicate that targeting this novel cancer-cell-initiated domino effect within the immune system–the gammadelta T cell/IL-17/neutrophil axis–represents a new strategy to inhibit metastatic disease.
-
Coffelt, S. B., et al (2015). "IL-17-producing gammadelta T cells and neutrophils conspire to promote breast cancer metastasis" Nature 522(7556): 345-348.
PubMed
Metastatic disease remains the primary cause of death for patients with breast cancer. The different steps of the metastatic cascade rely on reciprocal interactions between cancer cells and their microenvironment. Within this local microenvironment and in distant organs, immune cells and their mediators are known to facilitate metastasis formation. However, the precise contribution of tumour-induced systemic inflammation to metastasis and the mechanisms regulating systemic inflammation are poorly understood. Here we show that tumours maximize their chance of metastasizing by evoking a systemic inflammatory cascade in mouse models of spontaneous breast cancer metastasis. We mechanistically demonstrate that interleukin (IL)-1beta elicits IL-17 expression from gamma delta (gammadelta) T cells, resulting in systemic, granulocyte colony-stimulating factor (G-CSF)-dependent expansion and polarization of neutrophils in mice bearing mammary tumours. Tumour-induced neutrophils acquire the ability to suppress cytotoxic T lymphocytes carrying the CD8 antigen, which limit the establishment of metastases. Neutralization of IL-17 or G-CSF and absence of gammadelta T cells prevents neutrophil accumulation and downregulates the T-cell-suppressive phenotype of neutrophils. Moreover, the absence of gammadelta T cells or neutrophils profoundly reduces pulmonary and lymph node metastases without influencing primary tumour progression. Our data indicate that targeting this novel cancer-cell-initiated domino effect within the immune system–the gammadelta T cell/IL-17/neutrophil axis–represents a new strategy to inhibit metastatic disease.
-
Yamada, D. H., et al (2015). "Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection" Immunity 42(2): 379-390.
PubMed
Understanding how viruses subvert host immunity and persist is essential for developing strategies to eliminate infection. T cell exhaustion during chronic viral infection is well described, but effects on antibody-mediated effector activity are unclear. Herein, we show that increased amounts of immune complexes generated in mice persistently infected with lymphocytic choriomeningitis virus (LCMV) suppressed multiple Fcgamma-receptor (FcgammaR) functions. The high amounts of immune complexes suppressed antibody-mediated cell depletion, therapeutic antibody-killing of LCMV infected cells and human CD20-expressing tumors, as well as reduced immune complex-mediated cross-presentation to T cells. Suppression of FcgammaR activity was not due to inhibitory FcgammaRs or high concentrations of free antibody, and proper FcgammaR functions were restored when persistently infected mice specifically lacked immune complexes. Thus, we identify a mechanism of immunosuppression during viral persistence with implications for understanding effective antibody activity aimed at pathogen control.
-
Kearl, T. J., et al (2013). "Programmed death receptor-1/programmed death receptor ligand-1 blockade after transient lymphodepletion to treat myeloma" J Immunol 190(11): 5620-5628.
PubMed
Early phase clinical trials targeting the programmed death receptor-1/ligand-1 (PD-1/PD-L1) pathway to overcome tumor-mediated immunosuppression have reported promising results for a variety of cancers. This pathway appears to play an important role in the failure of immune reactivity to malignant plasma cells in multiple myeloma patients, as the tumor cells express relatively high levels of PD-L1, and T cells show increased PD-1 expression. In the current study, we demonstrate that PD-1/PD-L1 blockade with a PD-L1-specific Ab elicits rejection of a murine myeloma when combined with lymphodepleting irradiation. This particular combined approach by itself has not previously been shown to be efficacious in other tumor models. The antitumor effect of lymphodepletion/anti-PD-L1 therapy was most robust when tumor Ag-experienced T cells were present either through cell transfer or survival after nonmyeloablative irradiation. In vivo depletion of CD4 or CD8 T cells completely eliminated antitumor efficacy of the lymphodepletion/anti-PD-L1 therapy, indicating that both T cell subsets are necessary for tumor rejection. Elimination of myeloma by T cells occurs relatively quickly as tumor cells in the bone marrow were nearly nondetectable by 5 d after the first anti-PD-L1 treatment, suggesting that antimyeloma reactivity is primarily mediated by preactivated T cells, rather than newly generated myeloma-reactive T cells. Anti-PD-L1 plus lymphodepletion failed to improve survival in two solid tumor models, but demonstrated significant efficacy in two hematologic malignancy models. In summary, our results support the clinical testing of lymphodepletion and PD-1/PD-L1 blockade as a novel approach for improving the survival of patients with multiple myeloma.
Product Citations
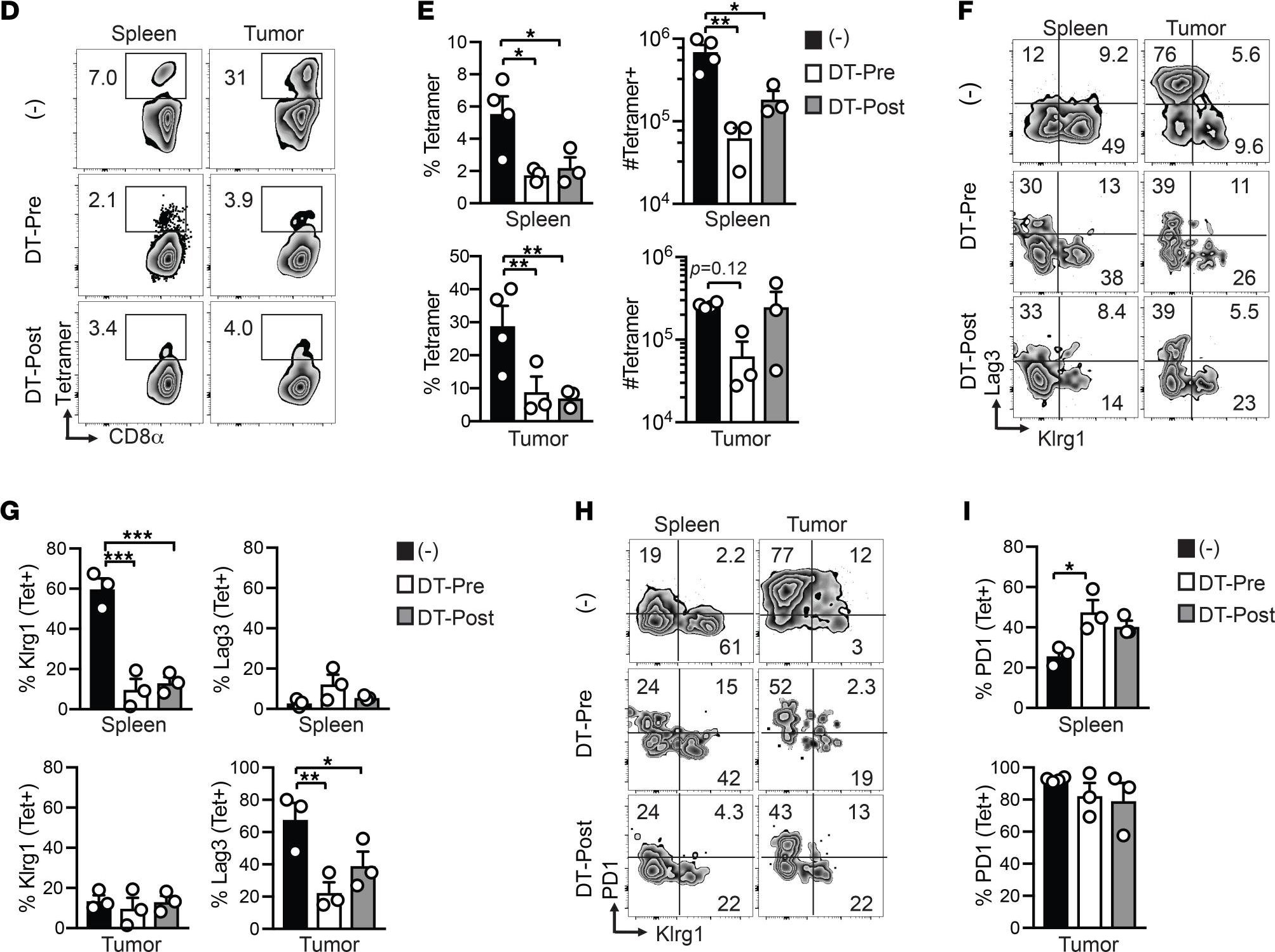
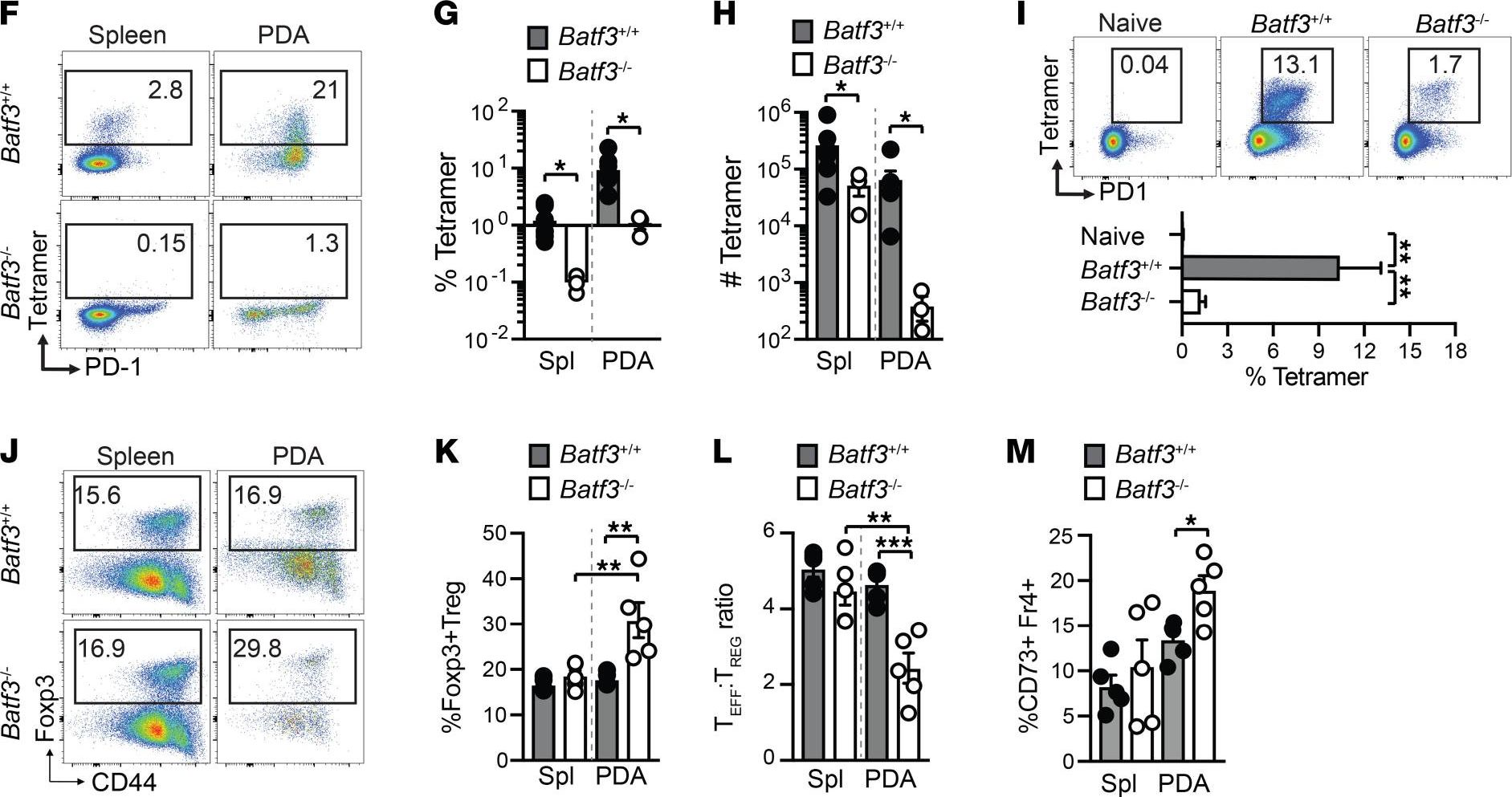
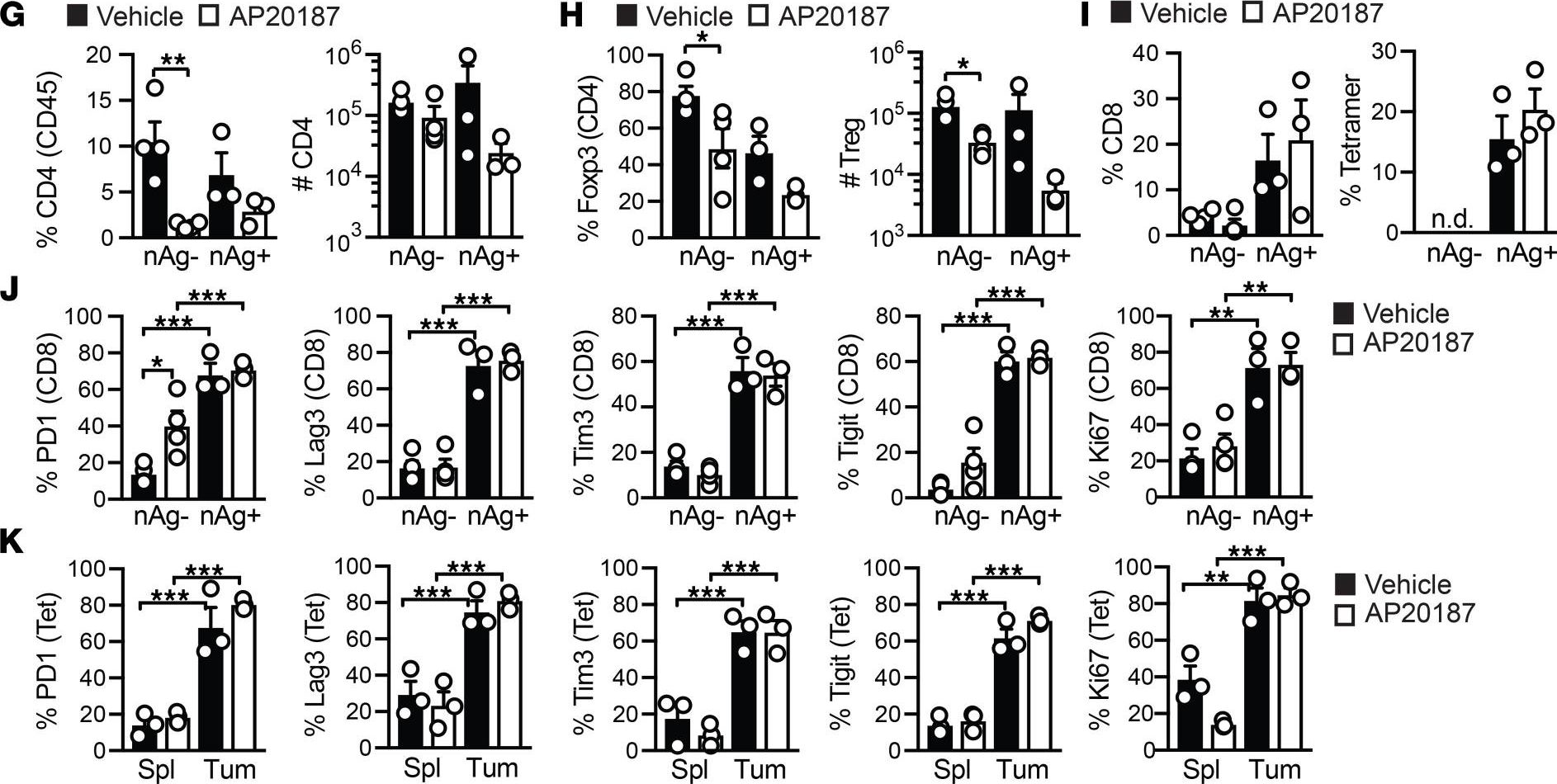
-
Microbiota-induced T cell plasticity enables immune-mediated tumour control.
In Nature on 1 March 2026 by Najar, T. A., Hao, Y., et al.
PubMed
Therapies that harness the immune system to target and eliminate tumour cells have revolutionized cancer care. Immune checkpoint blockade (ICB), which boosts the anti-tumour immune response by inhibiting negative regulators of T cell activation1-3, is remarkably successful in a subset of cancer patients. Yet a significant proportion do not respond to treatment, emphasizing the need to understand factors influencing the therapeutic efficacy of ICB4-9. The gut microbiota, consisting of trillions of microorganisms residing in the gastrointestinal tract, has emerged as a critical determinant of immune function and response to cancer immunotherapy, with several studies demonstrating association of microbiota composition with clinical response10-16. However, a mechanistic understanding of how gut commensal bacteria influence the efficacy of ICB remains elusive. Here we use a gut commensal microorganism, segmented filamentous bacteria (SFB), which induces an antigen-specific T helper 17 (TH17) cell effector program in the small intestine lamina propria (SILP)17, to investigate how colonization with this microbe affects the efficacy of ICB in restraining distal growth of tumours sharing antigen with SFB. We find that anti-programmed cell death protein 1 (PD-1) treatment effectively inhibits the growth of implanted SFB antigen-expressing melanoma only if mice are colonized with SFB. Through T cell receptor (TCR) clonal lineage tracing, fate mapping and peptide-major histocompatability complex (MHC) tetramer staining, we identify tumour-associated SFB-specific T helper 1 (TH1)-like cells derived from the homeostatic TH17 cells induced by SFB colonization in the SILP. These gut-educated ex-TH17 cells produce high levels of the pro-inflammatory cytokines interferon (IFN)-γ and tumour necrosis factor (TNF) within the tumour microenvironment (TME), enhancing antigen presentation and promoting recruitment, expansion and effector functions of CD8+ tumour-infiltrating cytotoxic lymphocytes and thereby enabling anti-PD-1-mediated tumour control. Conditional ablation of SFB-induced IL-17A+CD4+ T cells, precursors of tumour-associated TH1-like cells, abolishes anti-PD-1-mediated tumour control and markedly impairs tumour-specific CD8+ T cell recruitment and effector function within the TME. Our data, as a proof of principle, define a cellular pathway by which a single, defined intestinal commensal imprints T cell plasticity that potentiates PD-1 blockade, and indicate targeted modulation of the microbiota as a strategy to broaden ICB efficacy.
-
PROTAC-based synthetic lethality strategy endogenously activates systemic STING to boost antitumor immunity.
In Sci Adv on 27 February 2026 by Liu, Y., Jiang, M., et al.
PubMed
Activation of the stimulator of interferon genes (STING) pathway drives natural killer (NK) cells and T cells to orchestrate multidimensional antitumor immune responses. While cytosolic DNA accumulation represents a superior endogenous strategy for STING activation, DNA repair machinery substantially constrains its immunogenic potential. Here, we propose a promising therapeutic strategy that leverages proteolysis-targeting chimera (PROTAC)-mediated degradation of PARP1 [poly(ADP-ribose) polymerase 1] and BRD4 (bromodomain-containing protein 4) to induce synthetic lethality, thereby disrupting DNA repair machinery that drives nuclear-to-cytosolic DNA leakage, surpassing the STING activation threshold to ignite cGAS-STING-mediated innate immunity. Our strategy demonstrates superior antitumor efficacy across multiple tumor models, eliciting robust CD8+ T cell- and NK cell-mediated immunity while suppressing pulmonary metastasis progression. This strategic integration of synthetic lethality with an immunogenic stress response establishes a previously unidentified paradigm for expanding broad applications by cGAS-STING-mediated innate immunity.
-
The integrated stress response promotes immune evasion through lipocalin 2.
In Nature on 18 February 2026 by Bossowski, J. P., Pillai, R., et al.
PubMed
Cancer cells activate the integrated stress response (ISR) to adapt to stress and resist therapy1. ISR signals converge on activating transcription factor 4 (ATF4), which controls cell-intrinsic transcriptional programs that are involved in metabolic adaptation, survival and growth2,3. However, whether the ISR-ATF4 axis influences anti-tumour immune responses remains mostly unknown. Here we show that loss of ATF4 decreases tumour progression considerably in immunocompetent mice, but not in immunocompromised ones, by enhancing T cell-dependent anti-cancer immune responses. An unbiased genetic screen of ATF4-regulated genes identifies lipocalin 2 (LCN2) as the principal ATF4-dependent effector that impairs anti-tumour immunity by favouring infiltration with immunosuppressive interstitial macrophages. Furthermore, we find that LCN2 promotes T cell exclusion and immune evasion in preclinical mouse models, and correlates with decreased T cell infiltration in patients with lung and pancreatic adenocarcinomas. Anti-LCN2 antibodies promote robust anti-tumour T cell responses in mouse models of aggressive solid tumours. Our study shows that the ATF4-LCN2 axis has a cell-extrinsic role in suppressing anti-cancer immunity, and could pave the way for an immunotherapy approach that targets LCN2.
-
Deficiency of lysosomal TMEM175 in myeloid macrophages exerts anti-tumor immunity via inflammasome and cross-presentation pathway.
In Nat Commun on 14 February 2026 by Zhang, Z., Li, X., et al.
PubMed
Discovering more targets is of great importance for developing alternative interventions for tumor therapy. The roles of transmembrane protein 175 (TMEM175) in neurodegeneration diseases have been reported, however its functions in tumor immune surveillance are not known. We show that TMEM175 conditional knockout in macrophages inhibits the tumor growth and metastasis through promoting the anti-tumor immunity in the tumor microenvironment (TME), including elevated M1-like polarization, reduced M2-like polarization, and facilitated recruitment and activation of T cells and nature killer cells (NKs). The anti-tumor immunity is abrogated by caspase-1 inhibitor VX-765, anti-IL-1β, and anti-IL-18. Tmem175-/- bone marrow-derived macrophages (BMDMs) show enhanced tumor antigen cross-presentation that is further strengthened by IL-1β and IL-18. NLRP3 is robustly elicited in Tmem175-/- BMDMs by the tumor cell debris through lysosomal permeabilization and cathepsin B leakage. Finally, Tmem175-/- mice are more responsive to anti-PD-1. Our works implies TMEM175 to be a potential target for immunotherapy.