InVivoPlus anti-mouse PD-1 (CD279)
Product Description
Specifications
| Isotype | Armenian hamster IgG |
|---|---|
| Recommended Isotype Control(s) | InVivoPlus polyclonal Armenian hamster IgG |
| Recommended Dilution Buffer | InVivoPure pH 6.5 Dilution Buffer |
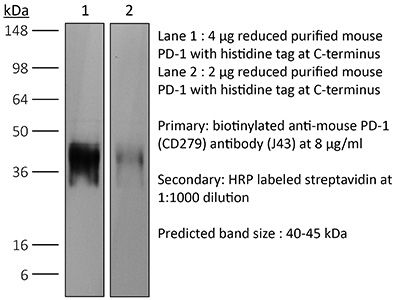
| Conjugation | This product is unconjugated. Conjugation is available via our Antibody Conjugation Services. |
| Immunogen | Syrian Hamster BKH cells transfected with mouse PD-1 cDNA |
| Reported Applications |
in vivo blocking of PD-1/PD-L signaling in vitro PD-1 neutralization Western blot |
| Formulation |
PBS, pH 6.5 Contains no stabilizers or preservatives |
| Endotoxin* |
≤0.5EU/mg (≤0.0005EU/μg) Determined by LAL assay |
| Aggregation* | <5%, Determined by SEC |
| Purity |
≥95% Determined by SDS-PAGE |
| Sterility | 0.2 µm filtration |
| Production | Purified from cell culture supernatant in an animal-free facility |
| Purification | Protein G |
| RRID | AB_1107747 |
| Molecular Weight | 150 kDa |
| Murine Pathogen Tests* |
Ectromelia/Mousepox Virus: Negative Hantavirus: Negative K Virus: Negative Lactate Dehydrogenase-Elevating Virus: Negative Lymphocytic Choriomeningitis virus: Negative Mouse Adenovirus: Negative Mouse Cytomegalovirus: Negative Mouse Hepatitis Virus: Negative Mouse Minute Virus: Negative Mouse Norovirus: Negative Mouse Parvovirus: Negative Mouse Rotavirus: Negative Mycoplasma Pulmonis: Negative Pneumonia Virus of Mice: Negative Polyoma Virus: Negative Reovirus Screen: Negative Sendai Virus: Negative Theiler’s Murine Encephalomyelitis: Negative |
| Storage | The antibody solution should be stored at the stock concentration at 4°C. Do not freeze. |
| Need a Custom Formulation? | See All Antibody Customization Options |
Application References
in vivo regulatory T cell depletion
in vivo blocking of PD-1/PD-L signaling
Sarraj, B., et al (2014). "Impaired selectin-dependent leukocyte recruitment induces T-cell exhaustion and prevents chronic allograft vasculopathy and rejection" Proc Natl Acad Sci U S A 111(33): 12145-12150.
PubMed
Selectin-selectin ligand interactions mediate the initial steps in leukocyte migration, an integral part of immune responses. Fucosyltransferase-VII (FucT-VII), encoded by Fut7, is essential for biosynthesis of selectin ligands. In an established model of cardiac allograft vasculopathy and chronic rejection, Fut7(-/-) recipients exhibited long-term graft survival with minimal vasculopathy compared with WT controls. Graft survival was associated with CD4 T-cell exhaustion in the periphery, characterized by impaired effector cytokine production, defective proliferation, increased expression of inhibitory receptors programmed death-1 (PD-1) and T cell Ig- and mucin-domain-containing molecule-3 (Tim-3), low levels of IL-7Ralpha on CD4 T cells, and reduced migration of polyfunctional CD4 memory T cells to the allograft. Blocking PD-1 triggered rejection only in Fut7(-/-) recipients, whereas depleting regulatory T cells had no effect in either Fut7(-/-) or WT recipients. Adoptive transfer experiments confirmed that this CD4 T cell-exhausted phenotype is seen primarily in Fut7(-/-) CD4 T cells. These data suggest that impaired leukocyte recruitment is a novel mechanism leading to CD4 T-cell exhaustion. Our experimental system serves as an excellent model to study CD4 T-cell exhaustion as a dominant mechanism of transplant tolerance. Further, targeting FucT-VII may serve as a promising strategy to prevent chronic allograft rejection and promote tolerance.
in vitro PD-1 neutralization
in vivo blocking of PD-1/PD-L signaling
in vitro PD-1 neutralization
Park, S. J., et al (2014). "Negative role of inducible PD-1 on survival of activated dendritic cells" J Leukoc Biol 95(4): 621-629.
PubMed
PD-1 is a well-established negative regulator of T cell responses by inhibiting proliferation and cytokine production of T cells via interaction with its ligands, B7-H1 (PD-L1) and B7-DC (PD-L2), expressed on non-T cells. Recently, PD-1 was found to be expressed in innate cells, including activated DCs, and plays roles in suppressing production of inflammatory cytokines. In this study, we demonstrate that PD-1 KO DCs exhibited prolonged longevity compared with WT DCs in the dLNs after transfer of DCs into hind footpads. Interestingly, upon LPS stimulation, WT DCs increased the expression of PD-1 and started to undergo apoptosis. DCs, in spleen of LPS-injected PD-1 KO mice, were more resistant to LPS-mediated apoptosis in vivo than WT controls. Moreover, treatment of blocking anti-PD-1 mAb during DC maturation resulted in enhanced DC survival, suggesting that PD-1:PD-L interactions are involved in DC apoptosis. As a result, PD-1-deficient DCs augmented T cell responses in terms of antigen-specific IFN-gamma production and proliferation of CD4 and CD8 T cells to a greater degree than WT DCs. Moreover, PD-1 KO DCs exhibited increased MAPK1 and CD40-CD40L signaling, suggesting a possible mechanism for enhanced DC survival in the absence of PD-1 expression. Taken together, our findings further extend the function of PD-1, which plays an important role in apoptosis of activated DCs and provides important implications for PD-1-mediated immune regulation.
Rabenstein, H., et al (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517.
PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
in vivo CTLA-4 neutralization
in vitro CTLA-4 neutralization
in vitro PD-1 neutralization
Noval Rivas, M., et al (2009). "Reviving function in CD4+ T cells adapted to persistent systemic antigen" J Immunol 183(7): 4284-4291.
PubMed
In bone marrow-transplanted patients, chronic graft-versus-host disease is a complication that results from the persistent stimulation of recipient minor histocompatibility Ag (mHA)-specific T cells contained within the graft. In this study, we developed a mouse model where persistent stimulation of donor T cells by recipient’s mHA led to multiorgan T cell infiltration. Exposure to systemic mHA, however, deeply modified T cell function and chronically stimulated T cells developed a long-lasting state of unresponsiveness, or immune adaptation, characterized by their inability to mediate organ immune damages in vivo. However, analysis of the gene expression profile of adapted CD4+ T cells revealed the specific coexpression of genes known to promote differentiation and function of Th1 effector cells as well as genes coding for proteins that control T cell activity, such as cell surface-negative costimulatory molecules and regulatory cytokines. Strikingly, blockade of negative costimulation abolished T cell adaptation and stimulated strong IFN-gamma production and severe multiorgan wasting disease. Negative costimulation was also shown to control lethal LPS-induced toxic shock in mice with adapted T cells, as well as the capacity of adapted T cells to reject skin graft. Our results demonstrate that negative costimulation is the molecular mechanism used by CD4+ T cells to adapt their activity in response to persistent antigenic stimulation. The effector function of CD4+ T cells that have adapted to chronic Ag presentation can be activated by stimuli strong enough to overcome regulatory signals delivered to the T cells by negative costimulation.
in vivo blocking of PD-1/PD-L signaling
in vivo CD8+ T cell depletion
Li, J., et al (2018). "Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells" Immunity 48(4): 773-786 e775.
PubMed
The molecular mechanisms whereby CD8(+) T cells become “exhausted” in the tumor microenvironment remain unclear. Programmed death ligand-1 (PD-L1) is upregulated on tumor cells and PD-1-PD-L1 blockade has significant efficacy in human tumors; however, most patients do not respond, suggesting additional mechanisms underlying T cell exhaustion. B7 superfamily member 1 (B7S1), also called B7-H4, B7x, or VTCN1, negatively regulates T cell activation. Here we show increased B7S1 expression on myeloid cells from human hepatocellular carcinoma correlated with CD8(+) T cell dysfunction. B7S1 inhibition suppressed development of murine tumors. Putative B7S1 receptor was co-expressed with PD-1 but not T cell immunoglobulin and mucin-domain containing-3 (Tim-3) at an activated state of early tumor-infiltrating CD8(+) T cells, and B7S1 promoted T cell exhaustion, possibly through Eomes overexpression. Combinatorial blockade of B7S1 and PD-1 synergistically enhanced anti-tumor immune responses. Collectively, B7S1 initiates dysfunction of tumor-infiltrating CD8(+) T cells and may be targeted for cancer immunotherapy.
in vivo blocking of PD-1/PD-L signaling
Imai, Y., et al (2015). "Cutting Edge: PD-1 Regulates Imiquimod-Induced Psoriasiform Dermatitis through Inhibition of IL-17A Expression by Innate gammadelta-Low T Cells" J Immunol 195(2): 421-425.
PubMed
Programmed cell death 1 (PD-1) is a key regulatory molecule that has been targeted in human cancers, including melanoma. In clinical testing, Abs against PD-1 have resulted in psoriasiform dermatitis (PsD). To determine whether PD-1 regulates PsD, we compared skin responses of PD-1-deficient (PD-1KO) mice and wild-type (WT) controls in an imiquimod (IMQ)-induced murine model of psoriasis. PD-1KO mice showed severe epidermal hyperplasia, greater neutrophilic infiltration, and higher expression of Th17 cytokines (versus WT mice). IMQ exposure increased PD-1 expression by skin gammadelta-low (GDL) T cells and enhanced expression of PD-L1 by keratinocytes. Three-fold increases in the percentage of IL-17A(+) GDL T cells were observed in skin cell suspensions derived from IMQ-treated PD-1KO mice (versus WT controls), suggesting that the lack of PD-1 has a functional effect not only on alphabeta T cells, but also on GDL T cells, and that PD-1 may play a regulatory role in PsD.
in vivo blocking of PD-1/PD-L signaling
Li, C., et al (2015). "ADAP and SKAP55 deficiency suppresses PD-1 expression in CD8+ cytotoxic T lymphocytes for enhanced anti-tumor immunotherapy" EMBO Mol Med 7(6): 754-769.
PubMed
PD-1 negatively regulates CD8(+) cytotoxic T lymphocytes (CTL) cytotoxicity and anti-tumor immunity. However, it is not fully understood how PD-1 expression on CD8(+) CTL is regulated during anti-tumor immunotherapy. In this study, we have identified that the ADAP-SKAP55 signaling module reduced CD8(+) CTL cytotoxicity and enhanced PD-1 expression in a Fyn-, Ca(2+)-, and NFATc1-dependent manner. In DC vaccine-based tumor prevention and therapeutic models, knockout of SKAP55 or ADAP showed a heightened protection from tumor formation or metastases in mice and reduced PD-1 expression in CD8(+) effector cells. Interestingly, CTLA-4 levels and the percentages of tumor infiltrating CD4(+)Foxp3(+) Tregs remained unchanged. Furthermore, adoptive transfer of SKAP55-deficient or ADAP-deficient CD8(+) CTLs significantly blocked tumor growth and increased anti-tumor immunity. Pretreatment of wild-type CD8(+) CTLs with the NFATc1 inhibitor CsA could also downregulate PD-1 expression and enhance anti-tumor therapeutic efficacy. Together, we propose that targeting the unrecognized ADAP-SKAP55-NFATc1-PD-1 pathway might increase efficacy of anti-tumor immunotherapy.
in vivo blocking of PD-1/PD-L signaling
in vivo CD8+ T cell depletion
Van der Jeught, K., et al (2014). "Intratumoral administration of mRNA encoding a fusokine consisting of IFN-beta and the ectodomain of the TGF-beta receptor II potentiates antitumor immunity" Oncotarget 5(20): 10100-10113.
PubMed
It is generally accepted that the success of immunotherapy depends on the presence of tumor-specific CD8(+) cytotoxic T cells and the modulation of the tumor environment. In this study, we validated mRNA encoding soluble factors as a tool to modulate the tumor microenvironment to potentiate infiltration of tumor-specific T cells. Intratumoral delivery of mRNA encoding a fusion protein consisting of interferon-beta and the ectodomain of the transforming growth factor-beta receptor II, referred to as Fbeta(2), showed therapeutic potential. The treatment efficacy was dependent on CD8(+) T cells and could be improved through blockade of PD-1/PD-L1 interactions. In vitro studies revealed that administration of Fbeta(2) to tumor cells resulted in a reduced proliferation and increased expression of MHC I but also PD-L1. Importantly, Fbeta(2) enhanced the antigen presenting capacity of dendritic cells, whilst reducing the suppressive activity of myeloid-derived suppressor cells. In conclusion, these data suggest that intratumoral delivery of mRNA encoding soluble proteins, such as Fbeta(2), can modulate the tumor microenvironment, leading to effective antitumor T cell responses, which can be further potentiated through combination therapy.
in vitro PD-1 neutralization
in vitro LAG-3 neutralization
in vitro blocking of IL-10R signaling
Verhagen, J. and D. C. Wraith (2014). "Blockade of LFA-1 augments in vitro differentiation of antigen-induced Foxp3(+) Treg cells" J Immunol Methods 414: 58-64.
PubMed
Adoptive transfer of antigen-specific, in vitro-induced Foxp3(+) Treg (iTreg) cells protects against autoimmune disease. To generate antigen-specific iTreg cells at high purity, however, remains a challenge. Whereas polyclonal T cell stimulation with anti-CD3 and anti-CD28 antibody yields Foxp3(+) iTreg cells at a purity of 90-95%, antigen-induced iTreg cells typically do not exceed a purity of 65-75%, even in a TCR-transgenic model. In a similar vein to thymic Treg cell selection, iTreg cell differentiation is influenced not only by antigen recognition and the availability of TGF-beta but also by co-factors including costimulation and adhesion molecules. In this study, we demonstrate that blockade of the T cell integrin Leukocyte Function-associated Antigen-1 (LFA-1) during antigen-mediated iTreg cell differentiation augments Foxp3 induction, leading to approximately 90% purity of Foxp3(+) iTreg cells. This increased efficacy not only boosts the yield of Foxp3(+) iTreg cells, it also reduces contamination with activated effector T cells, thus improving the safety of adoptive transfer immunotherapy.
in vitro PD-1 neutralization
Immunofluorescence
Schwager, K., et al (2013). "The immunocytokine L19-IL2 eradicates cancer when used in combination with CTLA-4 blockade or with L19-TNF" J Invest Dermatol 133(3): 751-758.
PubMed
Systemic high-dose IL2 promotes long-term survival in a subset of metastatic melanoma patients, but this treatment is accompanied by severe toxicities. The immunocytokine L19-IL2, in which IL2 is fused to the human L19 antibody capable of selective accumulation on tumor neovasculature, has recently shown encouraging clinical activity in patients with metastatic melanoma. In this study, we have investigated the therapeutic performance of L19-IL2, administered systemically in combination with a murine anti-CTLA-4 antibody or with a second clinical-stage immunocytokine (L19-TNF) in two syngeneic immunocompetent mouse models of cancer. We observed complete tumor eradications when L19-IL2 was used in combination with CTLA-4 blockade. Interestingly, mice cured from F9 tumors developed new lesions when rechallenged with tumor cells after therapy, whereas mice cured from CT26 tumors were resistant to tumor rechallenge. Similarly, L19-IL2 induced complete remissions when administered in a single intratumoral injection in combination with L19-TNF, whereas the two components did not lead to cures when administered as single agents. These findings provide a rationale for combination trials in melanoma, as the individual therapeutic agents have been extensively studied in clinical trials, and the antigen recognized by the L19 antibody has an identical sequence in mouse and man.
in vivo blocking of PD-1/PD-L signaling
Li, J., et al (2018). "Co-inhibitory Molecule B7 Superfamily Member 1 Expressed by Tumor-Infiltrating Myeloid Cells Induces Dysfunction of Anti-tumor CD8(+) T Cells" Immunity 48(4): 773-786 e775.
PubMed
The molecular mechanisms whereby CD8(+) T cells become “exhausted” in the tumor microenvironment remain unclear. Programmed death ligand-1 (PD-L1) is upregulated on tumor cells and PD-1-PD-L1 blockade has significant efficacy in human tumors; however, most patients do not respond, suggesting additional mechanisms underlying T cell exhaustion. B7 superfamily member 1 (B7S1), also called B7-H4, B7x, or VTCN1, negatively regulates T cell activation. Here we show increased B7S1 expression on myeloid cells from human hepatocellular carcinoma correlated with CD8(+) T cell dysfunction. B7S1 inhibition suppressed development of murine tumors. Putative B7S1 receptor was co-expressed with PD-1 but not T cell immunoglobulin and mucin-domain containing-3 (Tim-3) at an activated state of early tumor-infiltrating CD8(+) T cells, and B7S1 promoted T cell exhaustion, possibly through Eomes overexpression. Combinatorial blockade of B7S1 and PD-1 synergistically enhanced anti-tumor immune responses. Collectively, B7S1 initiates dysfunction of tumor-infiltrating CD8(+) T cells and may be targeted for cancer immunotherapy.
in vivo blocking of PD-1/PD-L signaling
Li, C., et al (2015). "ADAP and SKAP55 deficiency suppresses PD-1 expression in CD8+ cytotoxic T lymphocytes for enhanced anti-tumor immunotherapy" EMBO Mol Med 7(6): 754-769.
PubMed
PD-1 negatively regulates CD8(+) cytotoxic T lymphocytes (CTL) cytotoxicity and anti-tumor immunity. However, it is not fully understood how PD-1 expression on CD8(+) CTL is regulated during anti-tumor immunotherapy. In this study, we have identified that the ADAP-SKAP55 signaling module reduced CD8(+) CTL cytotoxicity and enhanced PD-1 expression in a Fyn-, Ca(2+)-, and NFATc1-dependent manner. In DC vaccine-based tumor prevention and therapeutic models, knockout of SKAP55 or ADAP showed a heightened protection from tumor formation or metastases in mice and reduced PD-1 expression in CD8(+) effector cells. Interestingly, CTLA-4 levels and the percentages of tumor infiltrating CD4(+)Foxp3(+) Tregs remained unchanged. Furthermore, adoptive transfer of SKAP55-deficient or ADAP-deficient CD8(+) CTLs significantly blocked tumor growth and increased anti-tumor immunity. Pretreatment of wild-type CD8(+) CTLs with the NFATc1 inhibitor CsA could also downregulate PD-1 expression and enhance anti-tumor therapeutic efficacy. Together, we propose that targeting the unrecognized ADAP-SKAP55-NFATc1-PD-1 pathway might increase efficacy of anti-tumor immunotherapy.
in vivo blocking of PD-1/PD-L signaling
Imai, Y., et al (2015). "Cutting Edge: PD-1 Regulates Imiquimod-Induced Psoriasiform Dermatitis through Inhibition of IL-17A Expression by Innate gammadelta-Low T Cells" J Immunol 195(2): 421-425.
PubMed
Programmed cell death 1 (PD-1) is a key regulatory molecule that has been targeted in human cancers, including melanoma. In clinical testing, Abs against PD-1 have resulted in psoriasiform dermatitis (PsD). To determine whether PD-1 regulates PsD, we compared skin responses of PD-1-deficient (PD-1KO) mice and wild-type (WT) controls in an imiquimod (IMQ)-induced murine model of psoriasis. PD-1KO mice showed severe epidermal hyperplasia, greater neutrophilic infiltration, and higher expression of Th17 cytokines (versus WT mice). IMQ exposure increased PD-1 expression by skin gammadelta-low (GDL) T cells and enhanced expression of PD-L1 by keratinocytes. Three-fold increases in the percentage of IL-17A(+) GDL T cells were observed in skin cell suspensions derived from IMQ-treated PD-1KO mice (versus WT controls), suggesting that the lack of PD-1 has a functional effect not only on alphabeta T cells, but also on GDL T cells, and that PD-1 may play a regulatory role in PsD.
in vivo blocking of PD-1/PD-L signaling
in vitro PD-1 neutralization
Park, S. J., et al (2014). "Negative role of inducible PD-1 on survival of activated dendritic cells" J Leukoc Biol 95(4): 621-629.
PubMed
PD-1 is a well-established negative regulator of T cell responses by inhibiting proliferation and cytokine production of T cells via interaction with its ligands, B7-H1 (PD-L1) and B7-DC (PD-L2), expressed on non-T cells. Recently, PD-1 was found to be expressed in innate cells, including activated DCs, and plays roles in suppressing production of inflammatory cytokines. In this study, we demonstrate that PD-1 KO DCs exhibited prolonged longevity compared with WT DCs in the dLNs after transfer of DCs into hind footpads. Interestingly, upon LPS stimulation, WT DCs increased the expression of PD-1 and started to undergo apoptosis. DCs, in spleen of LPS-injected PD-1 KO mice, were more resistant to LPS-mediated apoptosis in vivo than WT controls. Moreover, treatment of blocking anti-PD-1 mAb during DC maturation resulted in enhanced DC survival, suggesting that PD-1:PD-L interactions are involved in DC apoptosis. As a result, PD-1-deficient DCs augmented T cell responses in terms of antigen-specific IFN-gamma production and proliferation of CD4 and CD8 T cells to a greater degree than WT DCs. Moreover, PD-1 KO DCs exhibited increased MAPK1 and CD40-CD40L signaling, suggesting a possible mechanism for enhanced DC survival in the absence of PD-1 expression. Taken together, our findings further extend the function of PD-1, which plays an important role in apoptosis of activated DCs and provides important implications for PD-1-mediated immune regulation.
in vitro PD-1 neutralization
Verhagen, J. and D. C. Wraith (2014). "Blockade of LFA-1 augments in vitro differentiation of antigen-induced Foxp3(+) Treg cells" J Immunol Methods 414: 58-64.
PubMed
Adoptive transfer of antigen-specific, in vitro-induced Foxp3(+) Treg (iTreg) cells protects against autoimmune disease. To generate antigen-specific iTreg cells at high purity, however, remains a challenge. Whereas polyclonal T cell stimulation with anti-CD3 and anti-CD28 antibody yields Foxp3(+) iTreg cells at a purity of 90-95%, antigen-induced iTreg cells typically do not exceed a purity of 65-75%, even in a TCR-transgenic model. In a similar vein to thymic Treg cell selection, iTreg cell differentiation is influenced not only by antigen recognition and the availability of TGF-beta but also by co-factors including costimulation and adhesion molecules. In this study, we demonstrate that blockade of the T cell integrin Leukocyte Function-associated Antigen-1 (LFA-1) during antigen-mediated iTreg cell differentiation augments Foxp3 induction, leading to approximately 90% purity of Foxp3(+) iTreg cells. This increased efficacy not only boosts the yield of Foxp3(+) iTreg cells, it also reduces contamination with activated effector T cells, thus improving the safety of adoptive transfer immunotherapy.
in vivo blocking of PD-1/PD-L signaling
Van der Jeught, K., et al (2014). "Intratumoral administration of mRNA encoding a fusokine consisting of IFN-beta and the ectodomain of the TGF-beta receptor II potentiates antitumor immunity" Oncotarget 5(20): 10100-10113.
PubMed
It is generally accepted that the success of immunotherapy depends on the presence of tumor-specific CD8(+) cytotoxic T cells and the modulation of the tumor environment. In this study, we validated mRNA encoding soluble factors as a tool to modulate the tumor microenvironment to potentiate infiltration of tumor-specific T cells. Intratumoral delivery of mRNA encoding a fusion protein consisting of interferon-beta and the ectodomain of the transforming growth factor-beta receptor II, referred to as Fbeta(2), showed therapeutic potential. The treatment efficacy was dependent on CD8(+) T cells and could be improved through blockade of PD-1/PD-L1 interactions. In vitro studies revealed that administration of Fbeta(2) to tumor cells resulted in a reduced proliferation and increased expression of MHC I but also PD-L1. Importantly, Fbeta(2) enhanced the antigen presenting capacity of dendritic cells, whilst reducing the suppressive activity of myeloid-derived suppressor cells. In conclusion, these data suggest that intratumoral delivery of mRNA encoding soluble proteins, such as Fbeta(2), can modulate the tumor microenvironment, leading to effective antitumor T cell responses, which can be further potentiated through combination therapy.
in vivo blocking of PD-1/PD-L signaling
Sarraj, B., et al (2014). "Impaired selectin-dependent leukocyte recruitment induces T-cell exhaustion and prevents chronic allograft vasculopathy and rejection" Proc Natl Acad Sci U S A 111(33): 12145-12150.
PubMed
Selectin-selectin ligand interactions mediate the initial steps in leukocyte migration, an integral part of immune responses. Fucosyltransferase-VII (FucT-VII), encoded by Fut7, is essential for biosynthesis of selectin ligands. In an established model of cardiac allograft vasculopathy and chronic rejection, Fut7(-/-) recipients exhibited long-term graft survival with minimal vasculopathy compared with WT controls. Graft survival was associated with CD4 T-cell exhaustion in the periphery, characterized by impaired effector cytokine production, defective proliferation, increased expression of inhibitory receptors programmed death-1 (PD-1) and T cell Ig- and mucin-domain-containing molecule-3 (Tim-3), low levels of IL-7Ralpha on CD4 T cells, and reduced migration of polyfunctional CD4 memory T cells to the allograft. Blocking PD-1 triggered rejection only in Fut7(-/-) recipients, whereas depleting regulatory T cells had no effect in either Fut7(-/-) or WT recipients. Adoptive transfer experiments confirmed that this CD4 T cell-exhausted phenotype is seen primarily in Fut7(-/-) CD4 T cells. These data suggest that impaired leukocyte recruitment is a novel mechanism leading to CD4 T-cell exhaustion. Our experimental system serves as an excellent model to study CD4 T-cell exhaustion as a dominant mechanism of transplant tolerance. Further, targeting FucT-VII may serve as a promising strategy to prevent chronic allograft rejection and promote tolerance.
in vivo blocking of PD-1/PD-L signaling
Rabenstein, H., et al (2014). "Differential kinetics of antigen dependency of CD4+ and CD8+ T cells" J Immunol 192(8): 3507-3517.
PubMed
Ag recognition via the TCR is necessary for the expansion of specific T cells that then contribute to adaptive immunity as effector and memory cells. Because CD4+ and CD8+ T cells differ in terms of their priming APCs and MHC ligands we compared their requirements of Ag persistence during their expansion phase side by side. Proliferation and effector differentiation of TCR transgenic and polyclonal mouse T cells were thus analyzed after transient and continuous TCR signals. Following equally strong stimulation, CD4+ T cell proliferation depended on prolonged Ag presence, whereas CD8+ T cells were able to divide and differentiate into effector cells despite discontinued Ag presentation. CD4+ T cell proliferation was neither affected by Th lineage or memory differentiation nor blocked by coinhibitory signals or missing inflammatory stimuli. Continued CD8+ T cell proliferation was truly independent of self-peptide/MHC-derived signals. The subset divergence was also illustrated by surprisingly broad transcriptional differences supporting a stronger propensity of CD8+ T cells to programmed expansion. These T cell data indicate an intrinsic difference between CD4+ and CD8+ T cells regarding the processing of TCR signals for proliferation. We also found that the presentation of a MHC class II-restricted peptide is more efficiently prolonged by dendritic cell activation in vivo than a class I bound one. In summary, our data demonstrate that CD4+ T cells require continuous stimulation for clonal expansion, whereas CD8+ T cells can divide following a much shorter TCR signal.
in vitro PD-1 neutralization
Schwager, K., et al (2013). "The immunocytokine L19-IL2 eradicates cancer when used in combination with CTLA-4 blockade or with L19-TNF" J Invest Dermatol 133(3): 751-758.
PubMed
Systemic high-dose IL2 promotes long-term survival in a subset of metastatic melanoma patients, but this treatment is accompanied by severe toxicities. The immunocytokine L19-IL2, in which IL2 is fused to the human L19 antibody capable of selective accumulation on tumor neovasculature, has recently shown encouraging clinical activity in patients with metastatic melanoma. In this study, we have investigated the therapeutic performance of L19-IL2, administered systemically in combination with a murine anti-CTLA-4 antibody or with a second clinical-stage immunocytokine (L19-TNF) in two syngeneic immunocompetent mouse models of cancer. We observed complete tumor eradications when L19-IL2 was used in combination with CTLA-4 blockade. Interestingly, mice cured from F9 tumors developed new lesions when rechallenged with tumor cells after therapy, whereas mice cured from CT26 tumors were resistant to tumor rechallenge. Similarly, L19-IL2 induced complete remissions when administered in a single intratumoral injection in combination with L19-TNF, whereas the two components did not lead to cures when administered as single agents. These findings provide a rationale for combination trials in melanoma, as the individual therapeutic agents have been extensively studied in clinical trials, and the antigen recognized by the L19 antibody has an identical sequence in mouse and man.
in vitro PD-1 neutralization
Noval Rivas, M., et al (2009). "Reviving function in CD4+ T cells adapted to persistent systemic antigen" J Immunol 183(7): 4284-4291.
PubMed
In bone marrow-transplanted patients, chronic graft-versus-host disease is a complication that results from the persistent stimulation of recipient minor histocompatibility Ag (mHA)-specific T cells contained within the graft. In this study, we developed a mouse model where persistent stimulation of donor T cells by recipient’s mHA led to multiorgan T cell infiltration. Exposure to systemic mHA, however, deeply modified T cell function and chronically stimulated T cells developed a long-lasting state of unresponsiveness, or immune adaptation, characterized by their inability to mediate organ immune damages in vivo. However, analysis of the gene expression profile of adapted CD4+ T cells revealed the specific coexpression of genes known to promote differentiation and function of Th1 effector cells as well as genes coding for proteins that control T cell activity, such as cell surface-negative costimulatory molecules and regulatory cytokines. Strikingly, blockade of negative costimulation abolished T cell adaptation and stimulated strong IFN-gamma production and severe multiorgan wasting disease. Negative costimulation was also shown to control lethal LPS-induced toxic shock in mice with adapted T cells, as well as the capacity of adapted T cells to reject skin graft. Our results demonstrate that negative costimulation is the molecular mechanism used by CD4+ T cells to adapt their activity in response to persistent antigenic stimulation. The effector function of CD4+ T cells that have adapted to chronic Ag presentation can be activated by stimuli strong enough to overcome regulatory signals delivered to the T cells by negative costimulation.
Product Citations
-
-
Cancer Research
WNT pathway inhibition sensitizes HAT1-high lung cancers to treatment with PD-1 inhibitors.
In Cancer Cell Int on 24 October 2025 by Yang, T., Xie, Z., et al.
PubMed
Immunotherapies change the paradigm of current pulmonary oncological clinics, although majority of patients fail to benefit from these treatment modalities. HAT1 overexpression is frequently diagnosed in lung cancer patients. Effective immunotherapeutic scheme remains to be determined for this portion of patients.
-
-
-
Cancer Research
Gut-derived Faecalibaculum rodentium exerts anti-cancer effects on colorectal cancer by modulating PDPN-CLEC-2 signaling pathway.
In mSystems on 19 August 2025 by Yang, X., Liu, M., et al.
PubMed
Dynamic changes in microbial composition are related to the progression of colorectal cancer (CRC); however, specific microbes with anti-CRC properties have not been identified. Here, we aimed to determine the effect of Faecalibaculum rodentium on CRC progression. In this study, the effect of F. rodentium was assessed in an azoxymethane (AOM)/dextran sulfate sodium salt (DSS)-induced CRC mouse model as well as in human CRC cell lines. Changes in anti-tumor immune responses were evaluated by flow cytometry. The functional metabolites produced by F. rodentium were identified by gas chromatography‒mass spectrometry (GC‒MS). The subcutaneously injected BALB/c mice with CT26 CRC cells were used to evaluate the effect of acetate produced by F. rodentium. We found that supplementation with F. rodentium suppressed CRC in an AOM/DSS-induced CRC mouse model as well as in the human CRC cell lines SW620 and HT-29. GC‒MS revealed that acetate is a functional tumor-suppressive metabolite produced by F. rodentium, and the tumor volume and tumor weight in the CRC model were inhibited by acetate administration. Mechanistically, F. rodentium produced acetate, which suppressed the expression of PDPN on CD8+ T cells and that of C-type lectin-like receptor 2 (CLEC-2) on tumor cells. As a consequence, CD8+ T-cell immunity was increased both in vivo and in vitro. In addition, we revealed that the acetate produced by F. rodentium can modulate podoplanin (PDPN)/CLEC-2/PI3K/AKT/mTOR signaling in CRC. In summary, F. rodentium produced acetate inhibits CRC by promoting CD8+ T-cell immunity and modulating the PDPN-CLEC-2 pathway. Our data indicate that F. rodentium is a potential probiotic for cancer prevention and treatment.IMPORTANCEThis study identified Faecalibaculum rodentium as a novel probiotic that suppresses colorectal cancer (CRC) via acetate, which enhances CD8+ T-cell immunity by blocking PDPN-CLEC-2 and induces tumor cell apoptosis through PI3K/AKT/mTOR pathway. These findings validated F. rodentium and acetate as dual-action therapeutic candidates through immune response activation and intrinsic tumor targeting by murine models and human CRC cell lines, thereby providing innovative strategies for CRC treatment.
-
-
-
Cancer Research
-
Endocrinology and Physiology
-
Immunology and Microbiology
Caerin 1.1/1.9-mediated antitumor immunity depends on IFNAR-Stat1 signalling of tumour infiltrating macrophage by autocrine IFNα and is enhanced by CD47 blockade.
In Sci Rep on 30 January 2025 by Li, J., Luo, Y., et al.
PubMed
Previously, we demonstrated that natural host-defence peptide caerin 1.1/caerin 1.9 (F1/F3) increases the efficacy of anti-PD-1 and therapeutic vaccine, in a HPV16 + TC-1 tumour model, but the anti-tumor mechanism of F1/F3 is still unclear. In this study, we explored the impact of F1/F3 on the tumor microenvironment in a transplanted B16 melanoma model, and further investigated the mechanism of action of F1/F3 using monoclonal antibodies to deplete relevant cells, gene knockout mice and flow cytometry. We show that F1/F3 is able to inhibit the growth of melanoma B16 tumour cells both in vitro and in vivo. Depletion of macrophages, blockade of IFNα receptor, and Stat1 inhibition each abolishes F1/F3-mediated antitumor responses. Subsequent analysis reveals that F1/F3 increases the tumour infiltration of inflammatory macrophages, upregulates the level of IFNα receptor, and promotes the secretion of IFNα by macrophages. Interestingly, F1/F3 upregulates CD47 level on tumour cells; and blocking CD47 increases F1/F3-mediated antitumor responses. Furthermore, F1/F3 intratumor injection, CD47 blockade, and therapeutic vaccination significantly increases the survival time of B16 tumour-bearing mice. These results indicate that F1/F3 may be effective to improve the efficacy of ICB and therapeutic vaccine-based immunotherapy for human epithelial cancers and warrants consideration for clinical trials.
-
-
-
Cell Biology
Suppressing PD-L1 Expression via AURKA Kinase Inhibition Enhances Natural Killer Cell-Mediated Cytotoxicity against Glioblastoma.
In Cells on 6 July 2024 by Nguyen, T. T. T., Gao, Q., et al.
PubMed
Immunotherapies have shown significant promise as an impactful strategy in cancer treatment. However, in glioblastoma multiforme (GBM), the most prevalent primary brain tumor in adults, these therapies have demonstrated lower efficacy than initially anticipated. Consequently, there is an urgent need for strategies to enhance the effectiveness of immune treatments. AURKA has been identified as a potential drug target for GBM treatment. An analysis of the GBM cell transcriptome following AURKA inhibition revealed a potential influence on the immune system. Our research revealed that AURKA influenced PD-L1 levels in various GBM model systems in vitro and in vivo. Disrupting AURKA function genetically led to reduced PD-L1 levels and increased MHC-I expression in both established and patient-derived xenograft GBM cultures. This process involved both transcriptional and non-transcriptional pathways, partly implicating GSK3β. Interfering with AURKA also enhanced NK-cell-mediated elimination of GBM by reducing PD-L1 expression, as evidenced in rescue experiments. Furthermore, using a mouse model that mimics GBM with patient-derived cells demonstrated that Alisertib decreased PD-L1 expression in living organisms. Combination therapy involving anti-PD-1 treatment and Alisertib significantly prolonged overall survival compared to vehicle treatment. These findings suggest that targeting AURKA could have therapeutic implications for modulating the immune environment within GBM cells.
-
-
-
Immunology and Microbiology
-
Endocrinology and Physiology
-
Cancer Research
Caerin 1.1/1.9-mediated antitumor immunity depends on IFNAR-Stat1 signalling of tumour infiltrating macrophage by autocrine of IFNα and is enhanced by CD47 blockade
In Research Square on 13 December 2023 by Li, J., Luo, Y., et al.
-
-
-
Cancer Research
Advanced Age in Humans and Mouse Models of Glioblastoma Show Decreased Survival from Extratumoral Influence.
In Clin Cancer Res on 1 December 2023 by Johnson, M. O., Bell, A., et al.
PubMed
Glioblastoma (GBM) is the most common aggressive primary malignant brain tumor in adults with a median age of onset of 68 to 70 years old. Although advanced age is often associated with poorer GBM patient survival, the predominant source(s) of maladaptive aging effects remains to be established. Here, we studied intratumoral and extratumoral relationships between adult patients with GBM and mice with brain tumors across the lifespan.
-
-
-
Cancer Research
ATM deficiency confers specific therapeutic vulnerabilities in bladder cancer.
In Sci Adv on 24 November 2023 by Zhou, Y., Börcsök, J., et al.
PubMed
Ataxia-telangiectasia mutated (ATM) plays a central role in the cellular response to DNA damage and ATM alterations are common in several tumor types including bladder cancer. However, the specific impact of ATM alterations on therapy response in bladder cancer is uncertain. Here, we combine preclinical modeling and clinical analyses to comprehensively define the impact of ATM alterations on bladder cancer. We show that ATM loss is sufficient to increase sensitivity to DNA-damaging agents including cisplatin and radiation. Furthermore, ATM loss drives sensitivity to DNA repair-targeted agents including poly(ADP-ribose) polymerase (PARP) and Ataxia telangiectasia and Rad3 related (ATR) inhibitors. ATM loss alters the immune microenvironment and improves anti-PD1 response in preclinical bladder models but is not associated with improved anti-PD1/PD-L1 response in clinical cohorts. Last, we show that ATM expression by immunohistochemistry is strongly correlated with response to chemoradiotherapy. Together, these data define a potential role for ATM as a predictive biomarker in bladder cancer.
-
-
-
Immunology and Microbiology
6-Mercaptopurine potently inhibits recruitment of SHP2 by phosphorylated PD-1 to inhibit PD-1 signalling and enhance T cell function.
In Immunology on 1 October 2023 by Liu, L., Lei, Y., et al.
PubMed
Antibody inhibitors that block PD-1/PD-L1 interaction have been approved for oncological clinics, yielding impressive treatment effects. Small molecules inhibiting PD-1 signalling are at various stages of development, given that small molecular drugs are expected to outperform protein drugs in several ways. Currently, a significant portion of these small molecular inhibitors achieve this purpose by binding to a limited region of the PD-L1 protein, thereby limiting the choice of chemical structures. Alternative strategies for developing small-molecular PD-1 inhibitors are urgently needed to broaden the choice of chemical structures. Here, we report that 6-mercaptopurine (6-MP) inhibits PD-1 signalling, activates T cell function in vitro and in vivo and shrinks tumours by activating cytotoxic T cells. Mechanistically, 6-MP potently inhibited PD-1 signalling by blocking the recruitment of SHP2 by PD-1. Considering that 6-MP is a chemotherapeutic agent already approved by the FDA for childhood leukaemia, our work revealed a novel anti-tumour mechanism for this drug and suggests that 6-MP warrants further clinical evaluation for other tumour types.
-
-
-
Cancer Research
-
Cell Biology
In vivo genome-wide CRISPR screening identifies ZNF24 as a negative NF-κB modulator in lung cancer.
In Cell Biosci on 1 December 2022 by Liu, L., Lei, Y., et al.
PubMed
Systemic identification of tumor suppressor genes (TSGs) and elucidation of their signaling provide a new angle for understanding of tumorigenesis, which is important for developing successful treatment for lung cancer patients. In our current work, we conducted an in vivo screen for lung cancer TSGs through CRISPR/Cas9 mediated knockout of genes at genome-wide scale. We found that ZNF24 was a potent and clinically relevant TSG of lung cancer. Ectopic expression of ZNF24 arrested lung cancer cells in S phase. Mechanistically, ZNF24 bound to promoter region of P65 to negatively regulate its transcription and thereby the signaling activity of NF-κB pathway. This signaling cascade is clinically relevant. Importantly, we found that combinational inhibition of KRAS, NF-κB, and PD-1 effectively shrank autochthonous KrasG12D/ZNF24-/- lung cancers in transgenic mouse model. Our current work thus revealed an important role played by loss of function of ZNF24 in lung tumorigenesis and shed new light in precision medicine for a portion of lung cancer patients.
-
-
-
Immunohistochemistry
-
Cancer Research
-
Immunology and Microbiology
Breast cancer cell-derived extracellular vesicles promote CD8+ T cell exhaustion via TGF-β type II receptor signaling.
In Nat Commun on 1 August 2022 by Xie, F., Zhou, X., et al.
PubMed
Cancer immunotherapies have shown clinical success in various types of tumors but the patient response rate is low, particularly in breast cancer. Here we report that malignant breast cancer cells can transfer active TGF-β type II receptor (TβRII) via tumor-derived extracellular vesicles (TEV) and thereby stimulate TGF-β signaling in recipient cells. Up-take of extracellular vesicle-TβRII (EV-TβRII) in low-grade tumor cells initiates epithelial-to-mesenchymal transition (EMT), thus reinforcing cancer stemness and increasing metastasis in intracardial xenograft and orthotopic transplantation models. EV-TβRII delivered as cargo to CD8+ T cells induces the activation of SMAD3 which we demonstrated to associate and cooperate with TCF1 transcription factor to impose CD8+ T cell exhaustion, resulting in failure of immunotherapy. The levels of TβRII+ circulating extracellular vesicles (crEV) appears to correlate with tumor burden, metastasis and patient survival, thereby serve as a non-invasive screening tool to detect malignant breast tumor stages. Thus, our findings not only identify a possible mechanism by which breast cancer cells can promote T cell exhaustion and dampen host anti-tumor immunity, but may also identify a target for immune therapy against the most devastating breast tumors.
-
-
-
Cancer Research
-
Immunology and Microbiology
A nanovaccine for antigen self-presentation and immunosuppression reversal as a personalized cancer immunotherapy strategy.
In Nat Nanotechnol on 1 May 2022 by Liu, C., Liu, X., et al.
PubMed
The strategy of combining a vaccine with immune checkpoint inhibitors has been widely investigated in cancer management, but the complete response rate for this strategy is still unresolved. We describe a genetically engineered cell membrane nanovesicle that integrates antigen self-presentation and immunosuppression reversal (ASPIRE) for cancer immunotherapy. The ASPIRE nanovaccine is derived from recombinant adenovirus-infected dendritic cells in which specific peptide-major histocompatibility complex class I (pMHC-I), anti-PD1 antibody and B7 co-stimulatory molecules are simultaneously anchored by a programmed process. ASPIRE can markedly improve antigen delivery to lymphoid organs and generate broad-spectrum T-cell responses that eliminate established tumours. This work presents a powerful vaccine formula that can directly activate both native T cells and exhausted T cells, and suggests a general strategy for personalized cancer immunotherapy.
-
-
-
Flow cytometry/Cell sorting
-
Biochemistry and Molecular biology
-
Cancer Research
-
Immunology and Microbiology
Carfilzomib modulates tumor microenvironment to potentiate immune checkpoint therapy for cancer.
In EMBO Mol Med on 11 January 2022 by Zhou, Q., Liang, J., et al.
PubMed
Impressive clinical benefit is seen in clinic with PD-1 inhibitors on portion of cancer patients. Yet, there remains an urgent need to develop effective synergizers to expand their clinical application. Tumor-associated macrophage (TAM), a type of M2-polarized macrophage, eliminates or suppresses T-cell-mediated anti-tumor responses. Transforming TAMs into M1 macrophages is an attractive strategy of anti-tumor therapy. Here, we conducted a high-throughput screening and found that Carfilzomib potently drove M2 macrophages to express M1 cytokines, phagocytose tumor cells, and present antigens to T cells. Mechanistically, Carfilzomib elicited unfolded protein response (UPR), activated IRE1α to recruit TRAF2, and activated NF-κB to transcribe genes encoding M1 markers in M2 macrophages. In vivo, Carfilzomib effectively rewired tumor microenvironment through reprogramming TAMs into M1-like macrophages and shrank autochthonous lung cancers in transgenic mouse model. More importantly, Carfilzomib synergized with PD-1 antibody to almost completely regress autochthonous lung cancers. Given the safety profiles of Carfilzomib in clinic, our work suggested a potentially immediate application of combinational treatment with Carfilzomib and PD-1 inhibitors for patients with solid tumors.
-
-
-
In vivo experiments
-
Cancer Research
-
Immunology and Microbiology
Countering the advert effects of lung cancer on the anticancer potential of dendritic cell populations reinstates sensitivity to anti-PD-1 therapy.
In PLoS One on 1 December 2021 by Brassard, J., Gill, M. E., et al.
PubMed
Lung cancer is the leading cause of cancer-related deaths. While the recent use of immune checkpoint inhibitors significantly improves patient outcomes, responsiveness remains restricted to a small proportion of patients. Conventional dendritic cells (DCs) play a major role in anticancer immunity. In mice, two subpopulations of DCs are found in the lung: DC2s (CD11b+Sirpα+) and DC1s (CD103+XCR1+), the latest specializing in the promotion of anticancer immune responses. However, the impact of lung cancer on DC populations and the consequent influence on the anticancer immune response remain poorly understood. To address this, DC populations were studied in murine models of Lewis Lung Carcinoma (LLC) and melanoma-induced lung metastasis (B16F10). We report that direct exposure to live or dead cancer cells impacts the capacity of DCs to differentiate into CD103+ DC1s, leading to profound alterations in CD103+ DC1 proportions in the lung. In addition, we observed the accumulation of CD103loCD11b+ DCs, which express DC2 markers IRF4 and Sirpα, high levels of T-cell inhibitory molecules PD-L1/2 and the regulatory molecule CD200. Finally, DC1s were injected in combination with an immune checkpoint inhibitor (anti-PD-1) in the B16F10 model of resistance to the anti-PD-1 immune checkpoint therapy; the co-injection restored sensitivity to immunotherapy. Thus, we demonstrate that lung tumor development leads to the accumulation of CD103loCD11b+ DCs with a regulatory potential combined with a reduced proportion of highly-specialized antitumor CD103+ DC1s, which could promote cancer growth. Additionally, promoting an anticancer DC signature could be an interesting therapeutic avenue to increase the efficacy of existing immune checkpoint inhibitors.
-
-
-
Immunology and Microbiology
Conditional PD-1/PD-L1 Probody Therapeutics Induce Comparable Antitumor Immunity but Reduced Systemic Toxicity Compared with Traditional Anti-PD-1/PD-L1 Agents.
In Cancer Immunol Res on 1 December 2021 by Assi, H. H., Wong, C., et al.
PubMed
Immune-checkpoint blockade has revolutionized cancer treatment. However, most patients do not respond to single-agent therapy. Combining checkpoint inhibitors with other immune-stimulating agents increases both efficacy and toxicity due to systemic T-cell activation. Protease-activatable antibody prodrugs, known as Probody therapeutics (Pb-Tx), localize antibody activity by attenuating capacity to bind antigen until protease activation in the tumor microenvironment. Herein, we show that systemic administration of anti-programmed cell death ligand 1 (anti-PD-L1) and anti-programmed cell death protein 1 (anti-PD-1) Pb-Tx to tumor-bearing mice elicited antitumor activity similar to that of traditional PD-1/PD-L1-targeted antibodies. Pb-Tx exhibited reduced systemic activity and an improved nonclinical safety profile, with markedly reduced target occupancy on peripheral T cells and reduced incidence of early-onset autoimmune diabetes in nonobese diabetic mice. Our results confirm that localized PD-1/PD-L1 inhibition by Pb-Tx can elicit robust antitumor immunity and minimize systemic immune-mediated toxicity. These data provide further preclinical rationale to support the ongoing development of the anti-PD-L1 Pb-Tx CX-072, which is currently in clinical trials.
-
-
-
Cancer Research
-
Immunology and Microbiology
Inhibition of the BTK-IDO-mTOR axis promotes differentiation of monocyte-lineage dendritic cells and enhances anti-tumor T cell immunity.
In Immunity on 12 October 2021 by Sharma, M. D., Pacholczyk, R., et al.
PubMed
Monocytic-lineage inflammatory Ly6c+CD103+ dendritic cells (DCs) promote antitumor immunity, but these DCs are infrequent in tumors, even upon chemotherapy. Here, we examined how targeting pathways that inhibit the differentiation of inflammatory myeloid cells affect antitumor immunity. Pharmacologic inhibition of Bruton's tyrosine kinase (BTK) and the tryptophan-degrading enzyme indoleamine 2,3-dioxygenase (IDO) or deletion of Btk or Ido1 allowed robust differentiation of inflammatory Ly6c+CD103+ DCs during chemotherapy, promoting antitumor T cell responses and inhibiting tumor growth. Immature Ly6c+c-kit+ precursor cells had epigenetic profiles similar to conventional DC precursors; deletion of Btk or Ido1 promoted differentiation of these cells. Mechanistically, a BTK-IDO axis inhibited a tryptophan-sensitive differentiation pathway driven by GATOR2 and mTORC1, and disruption of the GATOR2 in monocyte-lineage precursors prevented differentiation into inflammatory DCs in vivo. IDO-expressing DCs and monocytic cells were present across a range of human tumors. Thus, a BTK-IDO axis represses differentiation of inflammatory DCs during chemotherapy, with implications for targeted therapies.
-
-
-
In vivo experiments
-
Cancer Research
-
Immunology and Microbiology
-
In vivo experiments
BFAR coordinates TGFβ signaling to modulate Th9-mediated cancer immunotherapy.
In J Exp Med on 5 July 2021 by Pei, S., Huang, M., et al.
PubMed
TGFβ is essential for the generation of anti-tumor Th9 cells; on the other hand, it causes resistance against anti-tumor immunity. Despite recent progress, the underlying mechanism reconciling the double-edged effect of TGFβ signaling in Th9-mediated cancer immunotherapy remains elusive. Here, we find that TGFβ-induced down-regulation of bifunctional apoptosis regulator (BFAR) represents the key mechanism preventing the sustained activation of TGFβ signaling and thus impairing Th9 inducibility. Mechanistically, BFAR mediates K63-linked ubiquitination of TGFβR1 at K268, which is critical to activate TGFβ signaling. Thus, BFAR deficiency or K268R knock-in mutation suppresses TGFβR1 ubiquitination and Th9 differentiation, thereby inhibiting Th9-mediated cancer immunotherapy. More interestingly, BFAR-overexpressed Th9 cells exhibit promising therapeutic efficacy to curtail tumor growth and metastasis and promote the sensitivity of anti-PD-1-mediated checkpoint immunotherapy. Thus, our findings establish BFAR as a key TGFβ-regulated gene to fine-tune TGFβ signaling that causes Th9 induction insensitivity, and they highlight the translational potential of BFAR in promoting Th9-mediated cancer immunotherapy.
-
-
-
Cancer Research
-
Immunology and Microbiology
Eomes Impedes Durable Response to Tumor Immunotherapy by Inhibiting Stemness, Tissue Residency, and Promoting the Dysfunctional State of Intratumoral CD8+ T Cells.
In Front Cell Dev Biol on 9 February 2021 by Sun, R., Wu, Y., et al.
PubMed
Sustaining efficacious T cell-mediated antitumor immune responses in the tumor tissues is the key to the success of cancer immunotherapy. Current strategies leverage altering the signals T cells sense in the tumor microenvironment (TME). Checkpoint inhibitor-based approaches block inhibitory signals such as PD-1 whereas cytokine-based therapies increase the level of immune-stimulatory cytokines such as IL-2. Besides extrinsic signals, the genetic circuit within T cells also participates in determining the nature and trajectory of antitumor immune responses. Here, we showed that efficacy of the IL33-based tumor immunotherapy was greatly enhanced in mice with T cell-specific Eomes deficiency. Mechanistically, we demonstrated that Eomes deficient mice had diminished proportions of exhausted/dysfunctional CD8+ T cells but increased percentages of tissue resident and stem-like CD8+ T cells in the TME. In addition, the IFNγ+TCF1+ CD8+ T cell subset was markedly increased in the Eomes deficient mice. We further demonstrated that Eomes bound directly to the transcription regulatory regions of exhaustion and tissue residency genes. In contrast to its role in inhibiting T cell immune responses at the tumor site, Eomes promoted generation of central memory T cells in the peripheral lymphoid system and memory recall responses against tumor growth at a distal tissue site. Finally, we showed that Eomes deficiency in T cells also resulted in increased efficacy of PD-1-blockade tumor immunotherapy. In all, our study indicates that Eomes plays a critical role in restricting prolonged T cell-mediated antitumor immune responses in the TME whereas promoting adaptive immunity in peripheral lymphoid organs.
-
-
-
Cancer Research
-
Immunology and Microbiology
Tumor-Derived IL33 Promotes Tissue-Resident CD8+ T Cells and Is Required for Checkpoint Blockade Tumor Immunotherapy.
In Cancer Immunol Res on 1 November 2020 by Chen, L., Sun, R., et al.
PubMed
Immune checkpoint blockade (ICB) immunotherapy has revolutionized cancer treatment by prolonging overall survival of patients with cancer. Despite advances in the clinical setting, the immune cellular network in the tumor microenvironment (TME) that mediates such therapy is not well understood. IL33 is highly expressed in normal epithelial cells but downregulated in tumor cells in advanced carcinoma. Here, we showed that IL33 was induced in tumor cells after treatment with ICB such as CTL antigen-4 (CTLA-4) and programmed death-1 (PD-1) mAbs. ST2 signaling in nontumor cells, particularly CD8+ T cells, was critical for the antitumor efficacy of ICB immunotherapy. We demonstrated that tumor-derived IL33 was crucial for the antitumor efficacy of checkpoint inhibitors. Mechanistically, IL33 increased the accumulation and effector function of tumor-resident CD103+CD8+ T cells, and CD103 expression on CD8+ T cells was required for the antitumor efficacy of IL33. In addition, IL33 also increased the numbers of CD103+ dendritic cells (DC) in the TME and CD103+ DC were required for the antitumor effect of IL33 and accumulation of tumor-infiltrating CD8+ T cells. Combination of IL33 with CTLA-4 and PD-1 ICB further prolonged survival of tumor-bearing mice. Our study established that the "danger signal" IL33 was crucial for mediating ICB cancer therapy by promoting tumor-resident adaptive immune responses.
-
-
-
Immunology and Microbiology
-
Cancer Research
A nanoscale metal organic frameworks-based vaccine synergises with PD-1 blockade to potentiate anti-tumour immunity.
In Nat Commun on 31 July 2020 by Li, X., Wang, X., et al.
PubMed
Checkpoint blockade therapy has provided noteworthy benefits in multiple cancers in recent years; however, its clinical benefits remain confined to 10-40% of patients with extremely high costs. Here, we design an ultrafast, low-temperature, and universal self-assembly route to integrate immunology-associated large molecules into metal-organic-framework (MOF)-gated mesoporous silica (MS) as cancer vaccines. Core MS nanoparticles, acting as an intrinsic immunopotentiator, provide the niche, void, and space to accommodate antigens, soluble immunopotentiators, and so on, whereas the MOF gatekeeper protects the interiors from robust and off-target release. A combination of MOF-gated MS cancer vaccines with systemic programmed cell death 1 (PD-1) blockade therapy generates synergistic effects that potentiate antitumour immunity and reduce the effective dose of an anti-PD-1 antibody to as low as 1/10 of that for PD-1 blockade monotherapy in E.G7-OVA tumour-bearing mice, with eliciting the robust adaptive OVA-specific CD8+ T-cell responses, reversing the immunosuppressive pathway and inducing durable tumour suppression.
-
-
-
Biochemistry and Molecular biology
Blocking interaction between SHP2 and PD-1 denotes a novel opportunity for developing PD-1 inhibitors.
In EMBO Mol Med on 8 June 2020 by Fan, Z., Tian, Y., et al.
PubMed
Small molecular PD-1 inhibitors are lacking in current immuno-oncology clinic. PD-1/PD-L1 antibody inhibitors currently approved for clinical usage block interaction between PD-L1 and PD-1 to enhance cytotoxicity of CD8+ cytotoxic T lymphocyte (CTL). Whether other steps along the PD-1 signaling pathway can be targeted remains to be determined. Here, we report that methylene blue (MB), an FDA-approved chemical for treating methemoglobinemia, potently inhibits PD-1 signaling. MB enhances the cytotoxicity, activation, cell proliferation, and cytokine-secreting activity of CTL inhibited by PD-1. Mechanistically, MB blocks interaction between Y248-phosphorylated immunoreceptor tyrosine-based switch motif (ITSM) of human PD-1 and SHP2. MB enables activated CTL to shrink PD-L1 expressing tumor allografts and autochthonous lung cancers in a transgenic mouse model. MB also effectively counteracts the PD-1 signaling on human T cells isolated from peripheral blood of healthy donors. Thus, we identify an FDA-approved chemical capable of potently inhibiting the function of PD-1. Equally important, our work sheds light on a novel strategy to develop inhibitors targeting PD-1 signaling axis.
-